

Abstract
Therapeutic application of virus-based delivery systems often implies a change of the tropism of these vectors. This can be achieved by insertion of polypeptides (e.g., antibody fragments) in viral coat proteins. Such fusion proteins have only been used in viral vectors so far and, as part of a virus, they have not been available for a detailed biophysical characterization. We analyzed a fusion protein called VP1-Z, which is based on the polyoma virus coat protein VP1 and protein Z. Protein Z is an engineered antibody-binding domain derived from protein A from Staphylococcus aureus. The fusion VP1-Z was constructed by insertion of protein Z in the HI-loop of VP1. As wild-type VP1, VP1-Z formed pentameric capsomers and assembled to VLPs in vitro. The stability of these particles was very similar compared to that of VLPs of wild-type VP1. Protein Z was fully structured in the fusion protein and was still capable of binding antibodies on the surface of VLPs of VP1-Z. Using this fusion protein, we could change the tropism of polyoma VLPs toward cells presenting on their surface the antigen of the coupled antibody.
Keywords: Polyoma, VP1, protein Z, antibody, stability, targeting, fluorescence microscopy
Murine polyoma virus is a nonenveloped DNA virus that belongs to the family Papovaviridae. The viral coat consists of three coat proteins VP1, VP2, and VP3 (Eckhart 1990). However, the formation of VLPs does not depend on the presence of all three polyoma coat proteins. It is possible to form VLPs from isolated recombinant VP1 in vitro (Salunke et al. 1986, 1989; Garcea et al. 1987). Usually these VLPs, which are ∼50 nm in diameter, consist of 360 VP1 molecules arranged in 72 pentamers. However, depending on the conditions used for assembly, smaller particles that contain only 24 or 18 pentamers can be produced (Salunke et al. 1989).
VLPs of VP1 can be used to package DNA and they have successfully been applied in cell transfection experiments (Forstova et al. 1995; Soeda et al. 1998; Braun et al. 1999; Krauzewicz et al. 2000) thus showing their value as vector system for gene or cancer therapy. In the case of cell transfection, the adhesion of VP1 to the cell surface is mediated by its natural sialic acid-binding activity (Freund et al. 1991; Stehle and Harrison 1996, 1997). As for the presence of sialic acid residues on the surface of almost all mammalian cells, this specific binding leads to cell-type-unspecific targeting of VLPs, based on VP1.
To achieve cell-type specificity of this potentially useful vector system, two modifications have to be established: Suppression of the natural tropism of polyoma VP1 and generation of a new cell-type-specific targeting function. Several different approaches have been pursued to endow viral vector systems with targeting functions. One attempt is the insertion of single-chain Fv fragments, which are the smallest antigen-binding antibody fragments, consisting of only the variable domains VL and VH in viral coat proteins (Chu et al. 1994; Jiang et al. 1998; Konishi et al. 1998; Martin et al. 1998). However, this strategy is limited by the modification of the virus coat protein and the development and characterization of single-chain Fv fragments. Furthermore, the fusion of all parts to a stable fusion protein that has the desired properties is essential. Another strategy is the insertion of docking modules on the surface of the viral coat protein. This would allow one to work with a once modified and characterized viral coat protein in several different applications. Such an approach was developed by Ohno and coworkers (Ohno and Meruelo 1997; Ohno et al. 1997). They used an immunoglobulin-binding domain from Staphylococcus aureus, the protein Z, which was inserted in the viral coat protein of Sindbis virus and retroviruses. Binding of a cell-specific antibody to the modified virus led to a change in the tropism of Sindbis virus toward the target cells of the respective antibody.
We transferred this elegant approach to VLPs of polyoma VP1. VP1 is a pentameric protein with monomers of 42.5 kD molecular mass. The VP1 structure shows mainly β-sheets, which are arranged in a jelly-roll topology (Stehle et al. 1994; Stehle and Harrison 1996, 1997). The VP1 monomer possesses three protruding loops, which are faced outward in the assembled state of the VLPs. Recently, we showed that the 18-kD ecDHFR could be functionally inserted in the HI-loop of VP1 (Gleiter et al. 1999). Therefore, we decided to insert the sequence of the immunoglobulin-binding domain, the protein Z, into the HI-loop of VP1 (Fig. 1 ▶). With this docking module it should be possible to create a recombinant carrier for proteins or DNA, which could be targeted via a bound antibody to a specific cell surface. Here, we characterize the fusion protein VP1-Z biochemically, with emphasis on the structural and functional parameters of the fusion protein.
Fig. 1.

Scheme of the fusion protein VP1-Z. The model is composed of the structures of VP1 (PDB entry 1vps) and the protein Z (PDB entry 2spz). The serine–glycine linkers were introduced with the program SYBYLL. No extensive attempts were made to energy minimize the structure of the fusion protein.
Results
Expression and purification
Recombinant expression of the VP1-Z gene in Escherichia coli led to formation of predominantly soluble VP1-Z in the host cell, only ∼10–20 % of the recombinant protein formed insoluble aggregates (not shown). VP1-Z was purified as described in the Materials and Methods section. The homogeneity of the purified protein is documented in Figure 2 ▶. Only one minor contamination was copurified. As described previously for wtVP1 (Leavitt et al. 1985), western blot analyses proved that this is a degradation product of VP1-Z, which is still integrated in the oligomeric structure of VP1-Z (data not shown).
Fig. 2.

Purification of VP1-Z. Escherichia coli strain BL21(DE3) that contained the plasmid pVP1Z was treated as described. Along the purification scheme, aliquots were taken and analyzed by 12% SDS-PAGE. VP1-Z is marked by an arrow. (1) Molecular weight standard; (2) soluble cell lysate; (3) VP1-Z after S-Sepharose; (4) VP1-Z pentamers after gel filtration; (5) VLPs of VP1-Z after gel filtration.
Structural characterization of VP1-Z
To analyze whether protein Z is structured within the fusion protein, CD spectroscopy was used. As an α-helical protein, protein Z showed a strong signal in the far UV region (Fig. 3A ▶). The α-helical content of isolated protein Z was determined to be 57% by quantitative analysis of the CD spectrum, using the program CDNN (Bohm et al. 1992), which is in accordance with 54% α-helical content of the three helices that constitute the protein Z structure (Tashiro et al. 1997). To determine the helix content of the Z domain in VP1-Z, CD spectra of wtVP1, VP1-Z, and protein Z were compared.
Fig. 3.

Secondary structure of VP1-Z. CD spectra from wtVP1, VP1-Z, and protein Z. VP1 variants were measured in 50 mM Tris at pH 7.4, 200 mM NaCl, 5% (v/v) glycerol, 0.5 mM EDTA, and 1 mM DTT at concentrations of 0.423 mg/mL (wtVP1) and 0.38 mg/mL (VP1-Z), respectively. Protein Z at a protein concentration of 0.15 mg/mL was analyzed in 10 mM Na-phosphate, pH 6.8. (A) Far UV CD spectra of pentameric wtVP1 (filled circle), VP1-Z (open circle) and protein Z (triangles) measured from 250 nm to 195 nm. The CD signals were normalized to ΘMRW. (B) Comparison of the calculated CD spectrum of VP1-Z (solid line), that is the sum of wtVP1 (filled circle) and protein Z (triangles), with the measured spectrum of VP1-Z (open circle). The CD data are normalized to Θmolar.
The predominantly β-sheet containing VP1 possessed a rather small amplitude in the far UV region (Fig. 3A ▶). The CD spectrum of VP1-Z proved to be identical to the sum of spectra from wtVP1 and protein Z (Fig. 3B ▶). This clearly indicated that the three helices of protein Z were maintained in the fusion protein. Thus both proteins, VP1 and protein Z, possessed a secondary structure that was not significantly altered within the fusion protein.
The oligomerization state of VP1-Z depends on the correct fold of the VP1 part in the fusion protein. WtVP1 and several variants were found previously to form pentamers in solution (Leavitt et al. 1985; Salunke et al. 1986; Schmidt et al. 1999; Gleiter et al. 1999; Schmidt et al. 2000; Stubenrauch et al. 2000). This association state could also be verified for VP1-Z. Using analytical ultracentrifugation the sedimentation velocity of VP1-Z pentamers was determined with s(W,20) = 7.8 S (Fig. 4 ▶), which is similar to that of pentameric wtVP1 (sapp = 7.5 S; Salunke et al. 1986). From sedimentation equilibrium measurements the molecular mass was calculated to be 237 kD (data not shown), corresponding to a pentamer of VP1-Z with a theoretical molecular mass of Mr = 247 kD.
Fig. 4.

Analytical ultracentrifugation of VP1-Z pentamers. The sedimentation velocity of VP1-Z (30,000 rpm, 20°C) was determined at a protein concentration of 0.45 mg/mL. Scans were taken every 10 min (displayed are every 20 min). Quantitative analysis yielded a value of s(w,20) = 7.8 S.
Stability of VP1-Z
The insertion of peptides or proteins between position Asn293 and Tyr294 of VP1 results in decreased stability of either VP1 (Stubenrauch et al. 2000) or the inserted protein (Gleiter et al. 1999). To analyze the stability of VP1-Z, thermal denaturation measurements of the fusion protein were performed either measuring fluorescence or CD. As shown in Figure 5 ▶, the isolated protein Z did not possess significant changes in secondary structure up to a temperature of ∼63°C. Unfolding of pentameric wtVP1 started at ∼48°C. The following aggregation on denaturation led to an increase in the CD signal at higher temperatures. Pentameric VP1-Z unfolded at ∼38°C, a significantly lower temperature than for denaturation of wtVP1. In contrast to wtVP1, the CD signal of VP1-Z reached a plateau during thermal denaturation. Denaturation of the Z-domain of VP1-Z was observed only at ∼62°C, which is similar to that of the isolated protein Z. Simultaneously, the fusion protein started to aggregate. Comparison of these data led to the conclusion that the VP1-part in the fusion protein is destabilized to some extent. Because all processes were irreversible, quantitative data could not be obtained. The Z domain in the fusion protein seemed to be as stable as the isolated protein Z.
Fig. 5.

Thermal denaturation of VP1-Z, wtVP1, and protein Z. Structural changes of VP1-Z (open circle), protein Z (triangles), and wtVP1 (filled circle) at protein concentrations of 200 μg/mL, respectively, in 20 mM Tris at pH 7.4, 5% glycerol, 200 mM NaCl, 0.5 mM EDTA were analyzed by CD at 220 nm and a heating rate of 1° C/min.
Functional analysis of VP1-Z
Considering the potential use of VP1-Z as the basic structure of a cell-type-specific delivery system, three functional aspects of VP1-Z are of major importance: The capability to assemble into VLPs, the inhibition of the natural sialic acid-binding activity, which leads to a cell-type-unspecific targeting of wtVP1 VLPs, and the binding of, for example, tumor-specific antibodies to protein Z on the surface of VLPs of VP1-Z.
The formation of VLPs was induced by dialysis of VP1-Z pentamers against assembly buffer. Sedimentation velocity analysis of assembled samples resulted in an S-value of sapp = 64 S (Fig. 6 ▶), which clearly indicates that an assembly process took place. The sizes of the VLPs obtained were comparable to those observed for other mutants of VP1 under similar conditions (Gleiter et al. 1999; Stubenrauch et al. 2000). The VLPs could also be visualized by electron microscopy after negative staining of VLP preparations (Fig. 6 ▶). Both analytical methods showed that the VLPs of VP1-Z pentamers were rather homogeneous in size, with a diameter of about 45 nm. Under identical conditions wtVP1 assembled predominantly to VLPs, comparable in size to those of VP1-Z (sapp = 63 S; diameter of ∼ 50 nm; Fig. 6 ▶). As shown by a less pronounced cooperativity in sedimentation, part of wtVP1 also assembled to larger particles (sapp = 96 S). This inhomogeneity might be caused by the respective buffer conditions, optimized for assembly of VP1-Z.
Fig. 6.
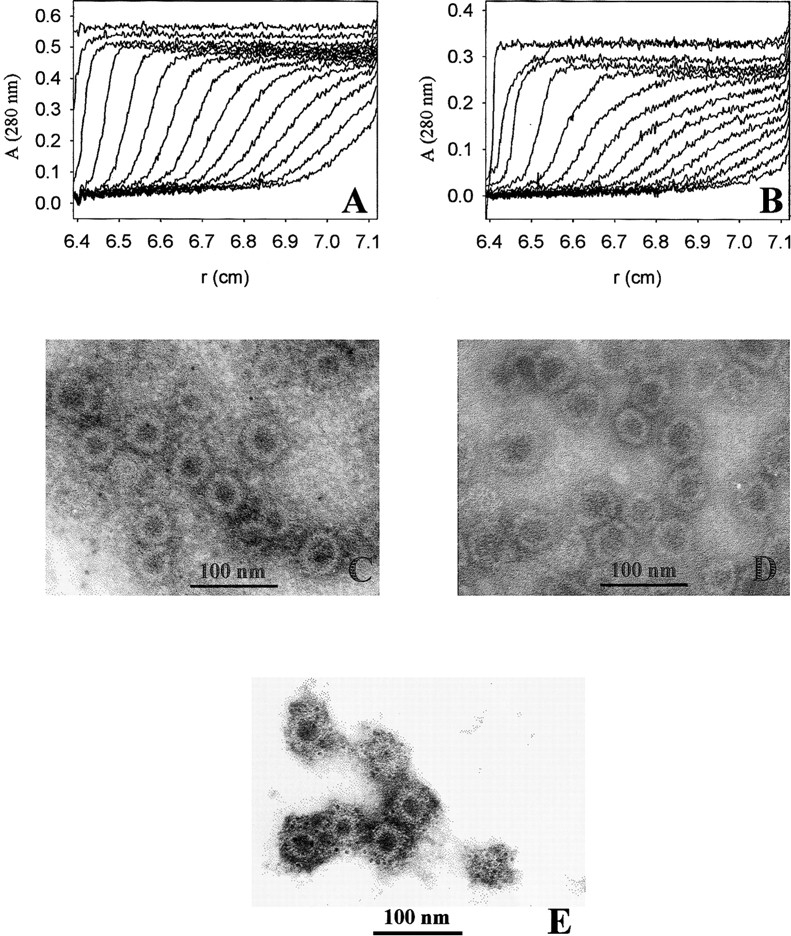
Formation of VLPs of VP1-Z. VP1-Z and wtVP1 at a protein concentration of ∼ 1 mg/mL was assembled to VLPs by dialysis against 20 mM Tris at pH 7.4, 200 mM NaCl, 5% glycerol, 0.5 M ammonium sulfate. The VLPs were analyzed by analytical ultracentrifugation. For electron microscopy the ammonium sulfate, which was present in the assembly buffer, was removed by dialysis. (A) Sedimentation velocity analysis of VP1-Z VLPs. An apparent S-value of 64 S was calculated. (B) Sedimentation velocity analysis of VLPs of wtVP1. The majority of the VLPs sedimented with sapp = 63 S. A small fraction assembled to larger particles (sapp = 96 S). VLPs of VP1-Z (C) and wtVP1 (D) were adsorbed to formvar-coated copper grids. The negative staining was performed using uranyl acetate. The electron micrograph was taken at a nominal magnification of 85,000×. (E) VLPs of VP1-Z adsorbed on Ni grids were coupled with gold-labeled antibody TU2. Afterward the complexes were stained with uranyl acetate. The electron micrograph was taken at a nominal magnification of 85,000×.
The stability of VLPs of VP1-Z and wtVP1 was very similar. The transition of thermal denaturation of VLPs of VP1-Z, measured by fluorescence, had a midpoint of TM = 51°C, whereas VLPs of wtVP1 showed a TM = 54°C (Fig. 7 ▶). Compared to the stability of nonassembled proteins, the VP1 variants are significantly stabilized on assembly, most likely because of additional interactions among the VP1 pentamers in the VLPs.
Fig. 7.

Stability of VLPs of VP1-Z and wtVP1 VP1-Z and wtVP1 at a protein concentration of 20 μg/mL in assembly buffer without ammonium sulfate, respectively, were incubated at a heating rate of 0.3°C/min from 5°C to 80°C. The change in fluorescence during heating was monitored using 295 nm for excitation and 340 nm for emission. The transition midpoint of denaturation of VP1-Z VLPs (open circle) and wtVP1 VLPs (filled circle) was determined with 51°C and 54°C, respectively.
Another ability of wtVP1 is its binding to sialic acid residues, which are located on the surface of most mammalian cells. The sialic acid-binding site of VP1 is located directly beneath the HI-loop (Stehle and Harrison 1997). As described previously, it is possible to diminish the binding of VP1 to its cellular target by the insertion of a polypeptide sequence in the HI-loop (Gleiter et al. 1999; Stubenrauch et al. 2000). In a hemagglutination assay, VLPs of wtVP1 induced agglutination of sheep erythrocytes at protein concentrations of ≥0.05 μg/mL. In contrast, the presence of VP1-Z did not lead to agglutination even at protein concentrations of ≤50 μg/mL (Fig. 8 ▶), which proves that the insertion of protein Z in the HI-loop of VP1 blocks binding of the natural ligand sialic acid to the protein. This is an essential prerequisite for establishing cell-type-specific targeting of modified polyoma VLPs. It should be mentioned, however, that the lack of hemagglutination does not prove the inability of VP1-Z to bind to any other cell. It is known that wtVP1 can bind sialyloligosaccharides in their branched and nonbranched forms, which seems to determine the pathogeneity of polyoma viruses in mice (Bauer et al. 1999). Whether the insertion of protein Z directly above the sialic acid binding site of VP1 not only reduces its affinity to the respective sialyloligosaccharides but also changes the specificity toward sugar residues, which are not present on erythrocytes, cannot be estimated from the present data.
Fig. 8.

Suppression of cell-type-unspecific cell adhesion of VP1-Z. Hemagglutination of sheep erythrocytes in the presence of VLPs of VP1-Z and wtVP1 was analyzed. Protein concentrations ranged from 50 μg/mL to 5 pg/mL (10-fold dilution from left to right). Only wtVP1 agglutinated the erythrocytes at a protein concentration of ≥50 ng/mL.
To analyze the function of the inserted polypeptide, the binding of a humanized anti-carcino embryogenic antigen (CEA) antibody to VP1-Z was measured. This antibody bound isolated protein Z with a dissociation constant of KD = 10 nM as determined by surface plasmon resonance measurements (data not shown). To show binding of the antibody on the surface of VP1-Z, the antibody was labeled with 5-nm gold particles. This gold-labeled antibody was used to visualize the specific binding on the surface of VP1-Z VLPs by electron microscopy. As shown in Figure 6E ▶, the electron-dense, 5-nm gold particles coupled to the antibody were attached to the VLPs. These ordered structures of the gold-labeled antibody could not be observed in samples containing VLPs of wtVP1 (data not shown). Thus the anti-CEA antibody specifically associated with the Z domain within the fusion protein VP1-Z. Furthermore, this result directly showed that, as predicted, the Z –domain, and thus the bound antibody, are presented on the surface of the VLPs. Titration of the antibody to VLPs of VP1-Z that were analyzed by HPLC gel filtration revealed that almost every Z domain on the surface of VP1 bound an antibody molecule, which led to a stoichiometry of about 0.8 anti–CEA antibody molecules per VP1-Z monomer. Furthermore, the appearance of the titration curve without any detectable curvature but, instead, with the sharp bend at a stoichiometry of 0.8 indicates that protein Z possessed a high affinity to the antibody, even within the fusion protein (Fig. 9 ▶). In contrast, no interaction between antibodies and wtVP1 could be observed, showing the specificity of the coupling of antibodies to VP1-Z (data not shown).
Fig. 9.

Titration of VP1-Z VLPs with anti-CEA antibody. The concentration of VP1-Z capsids was held constant at 1.5 μM referred to as the monomer of VP1-Z. The anti-CEA antibody was titrated to VP1-Z from 0 μM to 7.5 μM. The samples were analyzed after overnight incubation at 20°C on HPLC gel filtration. The relative area of the antibody that was not bound to VP1-Z capsids was measured and plotted against the molar ratio of VP1-Z monomer to anti-CEA antibody.
These antibody-binding characteristics were not limited to the anti-CEA antibody. Another humanized antibody, Herceptin, could also be coupled to VLPs of VP1-Z with a similar stoichiometry (data not shown).
Cell-specific targeting of antibody presenting VLPs
The presentation of a cell-type-specific antibody on the surface of VP1-Z VLPs allowed us to analyze whether or not the respective antibody leads to a change in the tropism of the modified VLPs. The antibody Herceptin is known to bind selectively to the glycoprotein p185Her2, a member of the EGF receptor family, which is present on several different human tumor cells (De Potter 1994; Molland et al. 1996). Using this antibody, a cell-type-specific targeting of immunoliposomes could be shown (Kirpotin et al. 1997). We compared the targeting of VLPs of VP1-Z, unmodified and decorated with Herceptin, to SK-BR-3 cells that expressed the mutated EGF receptor. To this end, rhodamine-labeled plasmid DNA was packaged into the respective VLPs and incubated with the cells for 90 min. As analyzed by laser scanning microscopy, almost no cellular-associated rhodamine fluorescence could be observed using VLPs in the absence of antibody (data not shown). A still very weakly detectable fluorescence signal was also obtained if DNA alone or in combination with the antibody was used. In contrast, if Herceptin was bound to the VLPs, nearly all of the cells were fluorescence labeled and the quantity of fluorescence per cell showed that this targeting was highly efficient (Fig. 10A ▶). This specific targeting function could be confirmed using MCF-7 cells as another antigen presenting cell line (data not shown). Furthermore, the fluorescence signal could not only be detected on the surface of the cells but also inside the cells (Fig. 10B–G ▶), which indicates that an uptake of the fluorescence-labeled particles occurred. Using the cell line HT-29, which does not present the antigen, no VLP complexes were associated with the cells. The same negative result was observed when the cell line SK-BR-3 was addressed by VLPs coupled with an antibody that was not directed against these cells (data not shown). Thus VLPs of VP1-Z that are decorated with a cell-type-specific antibody can be used specifically to deliver these particles to cells that express the antigen on the surface and to transport them across the cytoplasmic membrane.
Fig. 10.
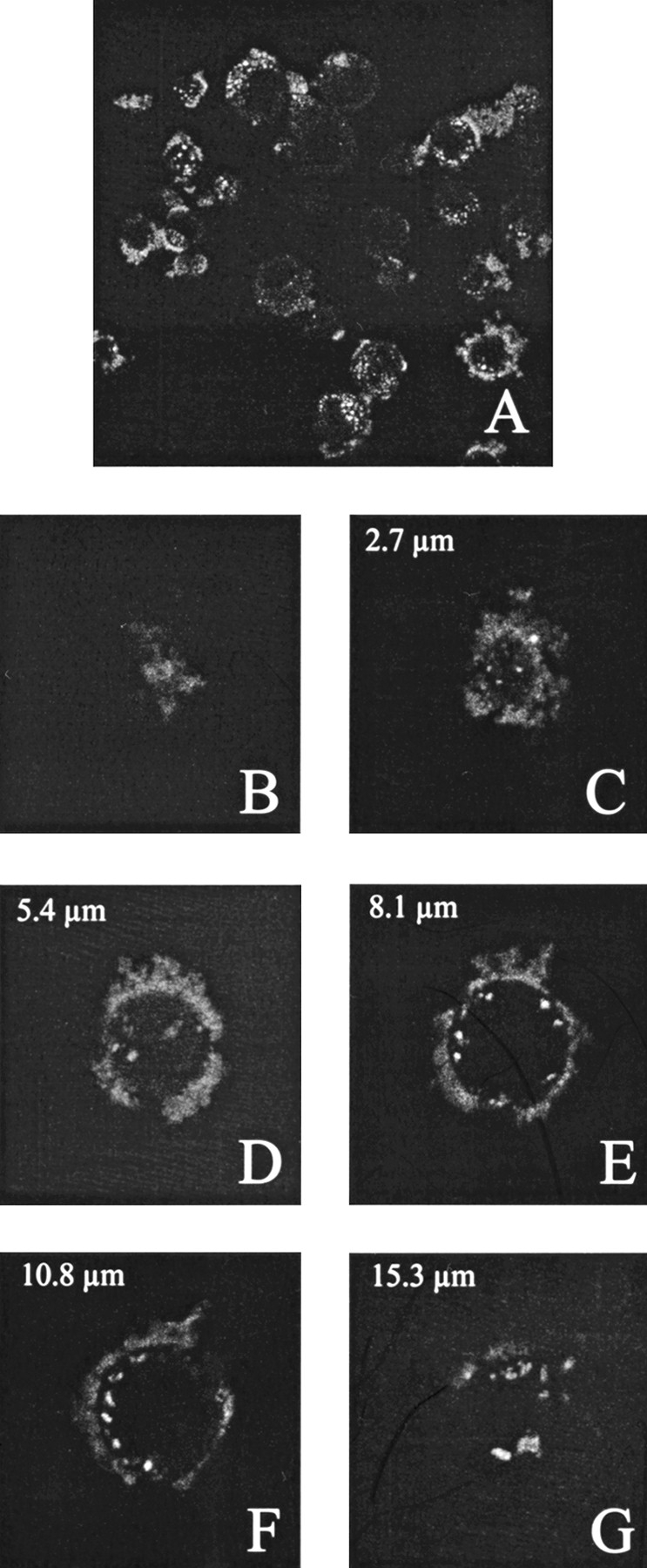
Cell type-specific targeting of antibody-presenting VLPs of VP1-Z. SK-BR-3 cells were grown to a confluency of ∼ 70%. Subsequently, the cells were incubated with VLPs of VP1-Z, which were decorated with the antibody Herceptin and packaged with fluorescence-labeled DNA. After overnight incubation nonbound VLP were removed by excessive washing and the cells were fixed using paraformaldehyde. (A) Overview of fluorescence-labeled cells. (B–G) Z series of a single cell. Fluorescence staining could be observed on the cell surface and inside the cells (D,E,F).
Discussion
Recently we showed that an 18-kDa protein can be functionally inserted in polyoma VP1 and presented on the surface of the respective VLPs. In this case, ecDHFR was used as a model system (Gleiter et al. 1999). Although some alterations regarding stability, enzymatic activity, and assembly of the VP1-DHFR fusion were observed compared with wtVP1 and ecDHFR, respectively, the results prompted us to go one step further in modifying the surface of VLPs of polyoma VP1 that should allow cell-type-specific targeting of those modified particles. To enable a change in tropism, the antibody-binding-domain protein Z was placed on the surface of VP1, thus making it possible to couple antibodies to VLPs of VP1-Z. The interaction of protein Z with antibodies is rather specific, with dissociation constants in the range of 15 nM (Starovasnik et al. 1997).
The inserted Z domain is rather small with a molecular mass of ∼6.8 kD. As the only secondary structure elements it consists of three helices, two of which are involved in antibody binding (Deisenhofer 1981). Importantly, the relative position of these helices does not change on binding of an antibody (Jendeberg et al. 1996), thus facilitating modeling of the fusion protein. We inserted protein Z via short serine–glycine linkers to enable an optimal flexibility on the surface of VP1. The structural integrity, not only of VP1 but also of protein Z, was completely maintained within the fusion protein, which led to a soluble, functional fusion protein. This was expected because protein Z is a derivative of the B-domain of protein A. Protein A consists of altogether five immunoglobulin-binding domains, which means that the B domain is flanked by other protein domains in protein A (Moks et al. 1986). Furthermore, there are several known artifical fusion proteins of protein Z (Chu et al. 1994; Martin et al. 1998), for example, the insertion of Z domain in Sindbis virus coat protein (Ohno and Meruelo 1997) or the use of a tandem Z domain as purification tag (Larsson et al. 1996) or as fusion partner to enhance solubility and refolding (Samuelsson et al. 1994; Samuelsson and Uhlen 1996). In all of these examples a functional fusion protein could be obtained.
The insertion of the model protein ecDHFR in VP1 at the same localization as that used for protein Z led to a dramatic destabilization of the DHFR part in the fusion protein (Gleiter et al. 1999). In contrast, protein Z does not seem to be significantly destabilized in the fusion protein compared with the stability of the isolated domain as revealed by CD measurements. Therefore, in contrast to DHFR, protein Z is still considerably more stable than VP1 within the fusion protein. VLPs of VP1-Z do not denature at temperatures <43°C, which indicates that the stability of these particles does not limit their potential as a gene therapeutic vector system at body temperature.
Whereas the apparent stability of VLPs of VP1-Z and wtVP1 is very similar, with a midpoint of thermal denaturation of TM = 51°C and TM = 54°C, respectively, the stability of the pentameric state of both proteins is quite different. Denaturation of wtVP1 occurs at 48°C. In contrast, the VP1-part of VP1-Z starts to denature at 38°C (Fig. 5A ▶). The observed stabilization, especially of VP1-Z in VLPs compared with VP1-Z pentamers, may result from two different types of additional interactions that occur on assembly. The C-terminal sequence of one molecule becomes a β-strand of the β-sheet of a VP1-Z molecule of a neighboring pentamer. Furthermore, the reduced VP1-Z is oxidized under assembly conditions. This oxidation process leads at least to one disulfide bridge between monomers within the pentamer (Schmidt et al. 2000).
The functional parameters of the fusion protein can be divided into three parts: The function of the VP1 part to assemble into VLPs, binding of antibody on the surface of VLPs, and suppression of the sialic acid binding on mammalian cells.
The assembly of VP1-Z into VLPs could be shown by different techniques. In all cases the data imply that the VLPs that are formed are a homogeneous population of particles with a diameter of ∼45 nm, which is very similar to particles of wtVP1 that are assembled under identical conditions. For wtVP1, particles of different sizes between 20 nm and 50 nm in diameter, depending on the assembly conditions, were described (Salunke et al. 1989). Once assembled, the different VLPs possess a similar stability against irreversible thermal denaturation.
The structural and functional integrity of the Z domain inserted in VP1 was proven by CD spectroscopy and antibody-binding activity. Quantitative analysis of the CD spectra clearly showed that protein Z, in solution and as fusion protein with VP1, consists of its natural α-helical content as described in the NMR structure (Tashiro et al. 1997; Gouda et al. 1998). In another study the crystal structure of protein Z was solved with only two of the three helices present (Deisenhofer 1981). However, this difference to the NMR structure of protein Z could be shown to be caused by crystal contacts of the protein in the crystal (Tashiro et al. 1997; Gouda et al. 1998).
Incubation of VLPs of VP1-Z with humanized monoclonal antibodies leads to a stoichiometry of binding of maximal 0.8, which indicates that almost every protein Z of the VLPs can bind one antibody molecule. Because of such a high loading rate, one cannot expect that every antigen-binding site of every bound antibody is accessible for the respective antigen. However, the titration clearly showed that the amount of antibody that is coupled can easily be controlled by loading substoichiometric amounts of antibody to VP1-Z. Although not analyzed so far, a low coupling rate would probably also ensure binding of different antibodies to VP1-Z simultaneously.
Placing cell-type-specific antibodies on the surface of VLPs, together with suppression of cell-type-unspecific sialic acid binding by the modification of VP1, should lead to a tropism of these particles based on the antibody—antigen interaction of the respective antibody. Such an antibody-specific cellular targeting could be shown using the tumor-specific antibody Herceptin. This antibody, directed against a mutant of a receptor of the EGF receptor family, directed the respective particles toward the antigen presenting cells SK-BR-3 or MCF-7. This targeting led also to an incorporation of these particles. Without the antibody coupled to the VLPs, no targeting could be observed at all. These results indicate that the modified polyoma VP1 particles that are described in this study are an important achievement in developing a cell-type-specific delivery system.
Materials and methods
Plasmid construction
The gene encoding the fusion protein VP1-Z was obtained by polymerase chain reaction (PCR) with three overlapping fragments: A, B, and C. Fragment A, which encodes residues 1–293 of VP1 and a C-terminal (Ser-Gly)3 linker sequence, was amplified from plasmid pVP1-DHFR (Gleiter et al. 1999) with primers NT7PpET (5′-CGATCCCGCGAAATTAATACGACTCAC-3′) and vp1linkrev (5′-ACCGCTACCGCTGCCGCTGTTTCTTGTAACTCTCCACC-3′). Fragment B encoded residues 294–385 of VP1, with an additional N-terminal (Ser-Gly)2-Ser linker sequence, and was amplified from the same plasmid with primers linkvp1fwd (5′TCTGGTTCTGGTAGCTATGATGTCCATCAC TGGAGAGG-3′) and revvp1not (5′- CCTAGGTCCAGCGGCC GCTCATTAATTTCCAGGAAATACAGTCTTTGTTTTTC-3′). Using this reverse primer, the last two amino acids of the VP1 sequence were changed to Gly384-Asn385, which is the natural C terminus of VP1. In addition, a NotI site was introduced. Fragment C consisted of the sequence encoding the Z domain amplified from plasmid pZR8 that comprised a C-terminal extended Z domain. Only the originally described sequence of protein Z (Larsson et al. 1996) was amplified from this plasmid with forward primer linkZfwd (5′- AGCGGCAGCGGTAGCGGTGTAGACAACAAA TTCAACAAAGAAC-3′) and the reverse primer zlinkrev (5′-GC TACCAGAACCAGATTTCGGCGCCTGAGCATCATTTAG-3′), thereby introducing the N- and C-terminal (Ser-Gly)-linker sequences, complementary to the C terminus of fragment A and to the N terminus of fragment B. Amplification products were separated on 1% agarose gels (ICN) and purified with the GFX purification kit (Amersham Pharmacia). Fragments B and C were combined for an overlapping amplification using primers linkZfwd and revvp1not.
The PCR product was again isolated from agarose gel and purified. This PCR product was used for the last overlapping PCR with fragment A and primers NT7PpET and revvp1not. The fragment was cloned into pET 21a(+; Novagen) using the restriction enzymes XbaI and NotI. The resulting plasmid, which contained the complete sequence of the fusion protein VP1-Z, was termed pVP1Z.
Protein expression
The expression strain BL21(DE3) was transformed with the plasmid pVP1Z. To optimize the expression conditions for the fusion protein cultivations were performed at different temperatures in 1 l shake flasks with 250 mL LB medium. At OD500 = 0.5 protein expression was induced by addition of IPTG to a final concentration of 1 mM. After induction, cells were incubated at temperatures between 15°C and 37°C overnight and then harvested by centrifugation. The pellet was resuspended in buffer A0 (50 mM Tris at pH 7.4, 200 mM NaCl, 5% (v/v) glycerol, 1 mM EDTA, 2 mM DTT). Cell lysis was achieved using a bead mill. Soluble and insoluble fractions were analyzed by SDS-PAGE.
For large-scale preparation of VP1-Z, E.coli BL21(DE3) transformed with pVP1Z was cultivated on mineral salt medium at 30°C at a scale of 5 L in a Biostat-Fermenter (B. Braun) using the fed-batch technique (Teich et al. 1998). The recombinant expression of VP1-Z was induced by adding 0.4 mM IPTG 3 hrs after feeding started. At the time of induction the temperature was set to 15°C and was maintained until the end of fermentation at 15 h. Cells were harvested by centrifugation (8000 g, 15 min) and stored at −70°C.
Protein purification
For preparation of VP1-Z 20 g cells (wet weight) were resuspended in 50-mL buffer A0, supplemented with two tablets of complete protease inhibitor cocktail, EDTA free (Roche Diagnostics). After high-pressure dispersion, the cell lysate was diluted with an equal volume of H20. To digest DNA bound to the N terminus of VP1-Z, the cell lysate was incubated with Benzonase (Merck) in the presence of 2 mM MgCl2 for 30 min at 4°C. Afterward EDTA was added to a final concentration of 4 mM and the lysate was centrifugated in a Sorvall SS-34 rotor at 20,000 rpm for 40 min.
The supernatant that contained the soluble fraction of VP1-Z was diluted with 50 mL buffer 1 (20 mM sodium acetate, pH 5.5, 5% (v/v) glycerol, 2 mM EDTA, 2 mM DTT) and the pH adjusted to 5.5. Because the pH shift led to slight aggregation the solution was again centrifugated (20,000 rpm, 40 min, SS-34 rotor) before the protein solution was applied to a S-Sepharose column (Amersham Pharmacia), equilibrated with buffer 1. The bound VP1-Z protein was eluted in a linear gradient from 0 to 2 M NaCl over 14 column volumes.
VP1-Z in the flow through was subjected to a second DNA digest after readjusting the pH and the Mg2+ concentration. Afterward the sample was applied again to the S-Sepharose column with the pH adjusted to 5.5. The VP1-Z containing fractions were pooled and concentrated by incubation with PEG 35,000 (Fluka) to a final volume of 6 mL for each run.
After dialysis against buffer A0 overnight VP1-Z was applied on a Superdex 200 prep grade gel filtration column (Amersham Pharmacia) equilibrated in buffer A0. Pentameric VP1-Z was concentrated and than dialyzed against VP1-Z assembly buffer (0.5 M ammonium sulfate, 20 mM Tris at pH 7.4, 5% (v/v) glycerol, 1 mM CaCl2) overnight at 20°C. The assembled VP1-Z again was applied on the Superdex 200 column, this time equilibrated in assembly buffer without ammonium sulfate (200 mM NaCl, 20 mM Tris at pH 7.4, 5% (v/v) glycerol, 1 mM CaCl2), thus separating VLPs and nonassembled pentamers. The overall yield of purified VP1-Z was in the range of 2 mg protein per 20 g of cells. Purification steps were analyzed on 12% SDS-PAGE, stained with Coomassie blue (Fairbanks et al. 1971).
The single IgG-binding domain of protein A (protein Z), which contained a fusion peptide of eight arginine residues and one cysteine at the C terminus of the protein, was also expressed in BL21(DE3). Cells expressing protein Z for about 14 hr were harvested and resuspended in buffer Z1 (10 mM phosphate, 10 mM EDTA, pH 6.8). After high-pressure disruption of the cells, the crude extract was centrifuged at 25,000 rpm for 1 hr. The clear supernatant was applied on a Resource S column (Amersham Pharmacia), which was equilibrated with buffer Z1. The recombinant protein was eluted in a linear gradient from 0–1 M NaCl. For further purification, the fractions containing protein Z were pooled and applied on a Superdex 75 column (Amersham Pharmacia), which was equilibrated in buffer Z1. Again, fractions containing protein Z were pooled and dialyzed against buffer Z1. For plasmon resonance measurements, the protein was dialyzed against buffer Z1 without EDTA.
Spectroscopic techniques
CD spectra were collected with an AVIV CD spectrophotometer. Protein concentrations for pentameric wtVP1 and VP1-Z were 0.423 mg/mL and 0.38 mg/mL, respectively. An average of 12 spectra were measured in the range of 250–195 nm in a 0.2-mm cuvette at 10°C.
The concentration of protein Z was 0.15 mg/mL in 10 mM phosphate buffer pH 6.8. For protein Z, an average of six spectra were measured in a 1-mm cuvette.
Thermal denaturation of all three proteins was measured in a Jasco CD spectrophotometer. The protein concentration for all proteins was 200 μg/mL. Measurements were performed in a 1-mm cuvette. The protein solution was heated using a programmable water bath with a heating rate of 1°C/min.
In addition, temperature transitions of the different proteins were determined using a Hitachi F-4500 fluorescence spectrometer in a stirrable cuvette with a light path of 10 mm. The excitation wavelength was set to 295 nm and the emission was measured at 340 nm. A programmable water bath was used to ensure a heating rate of about 0.3°C/min. Temperature transitions were measured in a range between 5°C and 80°C with protein concentrations of 20 μg/mL.
Analytical ultracentrifugation
VP1-Z at a concentration of ∼0.25 mg/mL in buffer A0 (pentamers) or assembly buffer (VLPs) was analyzed in a Beckman Optima XL-A centrifuge using an AN60Ti rotor and double sector cells. Sedimentation velocity experiments were performed at 30,000 rpm and 20°C for the pentameric state and at 10,000 rpm, 20°C in the case of VLPs, respectively. Sedimentation equilibrium centrifugation was performed at 10,000 rpm, 20°C. The data were analyzed using the software provided by Beckman Instruments.
Assembly of VP1-Z
Assembly of VP1-Z to VLPs was induced by dialyzing VP1-Z against assembly buffer (0. 5 M ammonium sulfate, 20 mM Tris at pH 7.4, 5% (v/v) glycerol, 1 mM CaCl2) overnight. After the end of assembly the ammonium sulfate was removed by dialysis against assembly buffer without ammonium sulfate (200 mM NaCl, 20 mM Tris at pH 7.4, 5% (v/v) glycerol, 1 mM CaCl2). After the second dialysis the samples were centrifuged at 13,000 rpm for 30 min at 4°C. Aliquots were used for electron microscopy and ultracentrifugation.
Association of VP1-Z with anti-CEA antibody
Because protein Z has highest affinity to the Fc part of human immunoglobulins of the type IgG, we used the monoclonal antibody MAK TU2. This antibody consists of a human Fc and Fab fragments from mouse, directed against the antigen CEA (Alfred Engel, Roche Diagnostics).
For association of VP1-Z capsids with anti-CEA antibody VP1-Z was used at a concentration of 1.5 μM (corresponding to the monomer). Variable concentrations of anti-CEA antibody (0 μM–7.5 μM) were incubated with VLPs of VP1-Z in assembly buffer without ammonium sulfate (200 mM NaCl, 20 mM Tris at pH 7.4, 5% (v/v) glycerol, 1 mM CaCl2). After incubation at 20°C overnight, the samples were applied to a YMC Diol-300 gel filtration column, which was equilibrated in the same buffer. The relative peak area of the remaining nonassociated antibody was analyzed.
Gold labeling of anti-CEA antibody
1-mL of gold solution (Sigma, 5-nm colloidal gold) adjusted to pH 7.3 with 0.2 M K2CO3 was incubated with 0.1 mL of anti-CEA solution (c = 0.6 mg/mL in 10 mM phosphate pH 6.8) for 30 min at room temperature. Afterward bovine serum albumin was added to a final concentration of 1%. Aggregates were sedimented by centrifugation for 1 hr at 30,000 rpm in a Beckman tabletop ultracentrifuge. The supernatant was discarded and the pellet was diluted with PBS to a final concentration of 0.1 mg/mL antibody–gold conjugate.
Electron microscopy
Formvar- and carbon-coated copper grids were used for negative staining of VLPs as described by Dyksta (1992) at protein concentrations of ∼0.2 mg/mL. Transmission electron microscopy was performed with a Zeiss EM 900. Photographs were taken at a nominal magnification of 85,000-fold.
Complexes of VLPs of VP1-Z and gold-labeled anti-CEA antibody were analyzed on formvar-coated Ni grids. VLPs with a concentration of ∼0.5 mg/mL were adsorbed to the grid for 30 min. Afterward the grid was blocked with 2% acetylated BSA and 0.2% Tween for 5 min. Finally, the grid was incubated for 10 min with gold-labeled antibody and then stained with uranyl acetate. Between each step excessive protein solution was carefully removed from the grid.
Hemagglutination assay
Hemagglutination of sheep erythrocytes (Dade-Behring) was determined in 0.9% (w/v) NaCl using U-well plates (NUNC). Protein concentrations in the assay ranged from 50 μg/mL to 0.5 × 10−9 μg/mL for both wtVP1 and VP1-Z, respectively. Fivefold dilutions of the protein solution were made in 0.9% NaCl. 40 μL of each dilution was supplemented with 40 μL of a 20-fold diluted erythrocyte stock solution, resulting in a 10-fold dilution per well. Hemagglutination was assayed after a 3-hr incubation at 7°C.
Cell targeting
The cells SK-BR-3 (ATCC: HTB-30) and MCF-7 (ATCC: HTB-22) were grown at 37°C in a humidified atmosphere, containing 5% CO2, using DMEM and RPMI 1640 medium, respectively. The medium was supplemented with 1% Glutamax (Gibco) and 10% heat-inactivated fetal calf serum (FCS). For targeting experiments, the cells were seeded in chamber slides, each chamber containing ∼70,000 cells. At a confluency of 70%, the targeting experiments were performed.
To analyze cellular targeting of the VLPs, 16 μg of VP1-Z or wtVP1 VLPs were incubated with 2 μg of rhodamine-labeled DNA of the plasmid pELI 92 (Yerushalmi et al. 2000; labeling procedure according to the manufacturer's guidelines [Mirus]). For specific targeting, 16 μg of MAK<Her2>rH-IgG (Herceptin) were added to the VLPs. The final sample volume was 150 μL of assembly buffer. Before adding the VLPs to the cells, the cells were supplemented with fresh, FCS-free medium. After at least 90-min incubation the cells were washed with PBS. For long-term storage the labeled cells were fixed with 4% (w/v) paraformaldehyde. Subsequently, mounting medium (Sigma) was added and the chamber slides were sealed.
Fluorescence microscopy was performed with a Zeiss LSM 410, equipped with a He/Ne-Laser. Samples were excitated at 543 nm and the emission filter was set to 570 nm. For scanning a single cell, Z series of 20 individual sectional planes were monitored with a pitch of 0.9 μM. Each plane was scanned four times to improve the signal-to-noise ratio.
Acknowledgments
We are grateful to S. Ståhl and M. Larsson for the kind gift of the plasmid encoding protein Z and to A. Engel and K.H. Sellinger (Roche Diagnostics GmbH) for the anti-CEA antibody TU-2 and Herceptin. G. Hause (Biozentrum, Universität Halle) is acknowledged for performing electron microscopy of the VLPs as is B. Hause for help with the LSM. We thank T. Jahn for technical assistance, F. Broda for help in modeling, and D. Wildemann for relief with the CD. R. Rudolph is acknowledged for fruitful discussions as is P. Bayer for critical reading of the manuscript. This work was supported by a grant of the Deutsche Forschungsgemeinschaft to H.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
DTT, dithiothreitol
ecDHFR, dihydrofolate reductase from Escherichia coli
EDTA, ethylenediaminetetraacetic acid
IPTG, isopropyl-β-D-thiogalactopyranoside
PDB, Protein Database Brookhaven
VLP, virus-like particle
VP1-Z, fusion protein with protein Z inserted in VP1
wt, wild type.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.31101.
References
- Bauer, P.H., Cui, C., Liu, R., Stehle, T., Harrison, S.C., DeCaprio, J.A., and Benjamin, T.L. 1999. Discrimination between sialic acid-containing receptors and pseudoreceptors regulates polyomavirus spread in the mouse. J. Virol. 73 5826–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm, G., Muhr, R., and Jaenicke, R. 1992. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 5 191–195. [DOI] [PubMed] [Google Scholar]
- Braun, H., Boller, K., Lower, J., Bertling, W.M., and Zimmer, A. 1999. Oligonucleotide and plasmid DNA packaging into polyoma VP1 virus-like particles expressed in Escherichia coli. Biotech. & App. Biochem. 29 Part 1 31–43. [PubMed] [Google Scholar]
- Chu, T.H., Martinez, I., Sheay, W.C., and Dornburg, R. 1994. Cell targeting with retroviral vector particles containing antibody-envelope fusion proteins. Gene Ther. 1 292–299. [PubMed] [Google Scholar]
- De Potter, C.R. 1994. The neu-oncogene: More than a prognostic indicator? Hum. Pathol. 25 1264–1268. [DOI] [PubMed] [Google Scholar]
- Deisenhofer, J. 1981. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9– and 2.8-A resolution. Biochemistry 20 2361–2370. [PubMed] [Google Scholar]
- Dyksta, M.J. 1992 Biological Electron Microscopy. Plenum Press, New York.
- Eckhart, W. 1990. Polyomavirinae and their replication. In Fundamental virology, (eds. B.N. Fields and D.M. Knipe), pp. 727–742. Raven Press, New York.
- Fairbanks, G., Steck, T., and Wallach, D. 1971. Electrophoretic analysis of the major polypeptides of the human erythrocyte membrane. Biochemistry 10 2606–2617. [DOI] [PubMed] [Google Scholar]
- Forstova, J., Krauzewicz, N., Sandig, V., Elliott, J., Palkova, Z., Strauss, M., and Griffin, B.E. 1995. Polyoma virus pseudocapsids as efficient carriers of heterologous DNA into mammalian cells. Hum.Gene Ther. 6 297–306. [DOI] [PubMed] [Google Scholar]
- Freund, R., Garcea, R.L., Sahli, R., and Benjamin, T.L. 1991. A single-amino-acid substitution in polyomavirus VP1 correlates with plaque size and hemagglutination behavior. J. Virol. 65 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcea, R.L., Salunke, D.M., and Caspar, D.L. 1987. Site-directed mutation affecting polyomavirus capsid self-assembly in vitro. Nature 329 86–87. [DOI] [PubMed] [Google Scholar]
- Gleiter, S., Stubenrauch, K., and Lilie, H. 1999. Changing the surface of a virus shell fusion of an enzyme to polyoma VP1. Protein Sci. 8 2562–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouda, H., Shiraishi, M., Takahashi, H., Kato, K., Torigoe, H., Arata, Y., and Shimada, I. 1998. NMR study of the interaction between the B domain of staphylococcal protein A and the Fc portion of immunoglobulin G. Biochemistry 37 129–136. [DOI] [PubMed] [Google Scholar]
- Jendeberg, L., Tashiro, M., Tejero, R., Lyons, B.A., Uhlen, M., Montelione, G.T., and Nilsson, B. 1996. The mechanism of binding staphylococcal protein A to immunoglobin G does not involve helix unwinding. Biochemistry 35 22–31. [DOI] [PubMed] [Google Scholar]
- Jiang, A., Chu, T.H., Nocken, F., Cichutek, K., and Dornburg, R. 1998. Cell-type-specific gene transfer into human cells with retroviral vectors that display single-chain antibodies. J. Virol. 72 10148–10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirpotin, D., Park, J.W., Hong, K., Zalipsky, S., Li, W.L., Carter, P., Benz, C.C., and Papahadjopoulos, D. 1997. Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry 36 66–75. [DOI] [PubMed] [Google Scholar]
- Konishi, H., Ochiya, T., Chester, K.A., Begent, R.H., Muto, T., Sugimura, T., Terada, M., and Begent, R.H. 1998. Targeting strategy for gene delivery to carcinoembryonic antigen-producing cancer cells by retrovirus displaying a single-chain variable fragment antibody. Hum.Gene Ther. 9 235–248. [DOI] [PubMed] [Google Scholar]
- Krauzewicz, N., Cox, C., Soeda, E., Clark, B., Rayner, S., and Griffin, B.E. 2000. Sustained ex vivo and in vivo transfer of a reporter gene using polyoma virus pseudocapsids. Gene Ther. 7 1094–1102. [DOI] [PubMed] [Google Scholar]
- Larsson, M., Brundell, E., Nordfors, L., Hoog, C., Uhlen, M., and Stahl, S. 1996. A general bacterial expression system for functional analysis of cDNA-encoded proteins. Protein Expr. Purif. 7 447–457. [DOI] [PubMed] [Google Scholar]
- Leavitt, A.D., Roberts, T.M., and Garcea, R.L. 1985. Polyoma virus major capsid protein, VP1. Purification after high level expression in Escherichia coli. J. Biol. Chem. 260 12803–12809. [PubMed] [Google Scholar]
- Martin, F., Kupsch, J., Takeuchi, Y., Russell, S., Cosset, F.L., and Collins, M. 1998. Retroviral vector targeting to melanoma cells by single-chain antibody incorporation in envelope. Hum. Gene Ther. 9 737–746. [DOI] [PubMed] [Google Scholar]
- Moks, T., Abrahmsen, L., Nilsson, B., Hellman, U., Sjoquist, J., and Uhlen, M. 1986. Staphylococcal protein A consists of five IgG-binding domains. Eur. J. Biochem. 156 637–643. [DOI] [PubMed] [Google Scholar]
- Molland, J.G., Barraclough, B.H., Gebski, V., Milliken, J., and Bilous, M. 1996. Prognostic significance of c-erbB-2 oncogene in axillary node-negative breast cancer. Aust. N.Z .J. Surg. 66 64–70. [DOI] [PubMed] [Google Scholar]
- Ohno, K. and Meruelo, D. 1997. Retrovirus vectors displaying the IgG-binding domain of protein A. Biochem. Mol. Med. 62 123–127. [DOI] [PubMed] [Google Scholar]
- Ohno, K., Sawai, K., Iijima, Y., Levin, B., and Meruelo, D. 1997. Cell-specific targeting of Sindbis virus vectors displaying IgG-binding domains of protein A. Nat. Biotechnol. 15 763–767. [DOI] [PubMed] [Google Scholar]
- Salunke, D.M., Caspar, D.L., and Garcea, R.L. 1986. Self-assembly of purified polyomavirus capsid protein VP1. Cell 46 895–904. [DOI] [PubMed] [Google Scholar]
- Salunke, D.M., Caspar, D.L., and Garcea, R.L. 1989. Polymorphism in the assembly of polyomavirus capsid protein VP1. Biophys. J. 56 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson, E., Moks, T., Nilsson, B., and Uhlen, M. 1994. Enhanced in vitro refolding of insulin-like growth factor I using a solubilizing fusion partner. Biochemistry 33 4207–4211. [DOI] [PubMed] [Google Scholar]
- Samuelsson, E. and Uhlen, M. 1996. Chaperone-like effect during in vitro refolding of insulin-like growth factor I using a solubilizing fusion partner. Ann. N.Y. Acad. Sci. 782 486–494. [DOI] [PubMed] [Google Scholar]
- Schmidt, U., Kenklies, J., Rudolph, R., and Bohm, G. 1999. Site-specific fluorescence labelling of recombinant polyomavirus-like particles. Biol. Chem. 380 397–401. [DOI] [PubMed] [Google Scholar]
- Schmidt, U., Rudolph, R., and Bohm, G. 2000. Mechanism of assembly of recombinant murine polyomavirus-like particles [In Process Citation]. J. Virol. 74 1658–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeda, E., Krauzewicz, N., Cox, C., Stokrova, J., Forstova, J., and Griffin, B.E. 1998. Enhancement by polylysine of transient, but not stable, expression of genes carried into cells by polyoma VP1 pseudocapsids. Gene Ther. 5 1410–1419. [DOI] [PubMed] [Google Scholar]
- Starovasnik, M.A., Braisted, A.C., and Wells, J.A. 1997. Structural mimicry of a native protein by a minimized binding domain. Proc. Natl. Acad. Sci. 94 10080–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle, T. and Harrison, S.C. 1996. Crystal structures of murine polyomavirus in complex with straight-chain and branched-chain sialyloligosaccharide receptor fragments. Structure 4 183–194. [DOI] [PubMed] [Google Scholar]
- Stehle, T. and Harrison, S.C. 1997. High-resolution structure of a polyomavirus VP1-oligosaccharide complex: implications for assembly and receptor binding. EMBO J. 16 5139–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle, T., Yan, Y., Benjamin, T.L., and Harrison, S.C. 1994. Structure of murine polyomavirus complexed with an oligosaccharide receptor fragment. Nature 369 160–163. [DOI] [PubMed] [Google Scholar]
- Stubenrauch, K., Bachmann, A., Rudolph, R., and Lilie, H. 2000. Purification of a viral coat protein by an engineered polyionic sequence. J. Chromatogr. B. Biomed. Sci. Appl. 737 77–84. [DOI] [PubMed] [Google Scholar]
- Tashiro, M., Tejero, R., Zimmerman, D.E., Celda, B., Nilsson, B., and Montelione, G.T. 1997. High-resolution solution NMR structure of the Z domain of staphylococcal protein A. J. Mol. Biol. 272 573–590. [DOI] [PubMed] [Google Scholar]
- Teich, A., Lin, H.Y., Andersson, L., Meyer, S., and Neubauer, P. 1998. Amplification of ColE1 related plasmids in recombinant cultures of Escherichia coli after IPTG induction. J. Biotechnol. 64 197–210. [DOI] [PubMed] [Google Scholar]
- Yerushalmi, N., Brinkmann, U., Brinkmann, E., Pai, L., and Pastan, I. 2000. Attenuating the growth of tumors by intratumoral administration of DNA encoding Pseudomonas exotoxin via cationic liposomes. Cancer Gene Ther. 7 91–96. [DOI] [PubMed] [Google Scholar]