

Abstract
Sequence similarity is the most common measure currently used to infer homology between proteins. Typically, homologous protein domains show sequence similarity over their entire lengths. Here we identify Asp box motifs, initially found as repeats in sialidases and neuraminidases, in new structural and sequence contexts. These motifs represent significantly similar sequences, localized to β hairpins within proteins that are otherwise different in sequence and three-dimensional structure. By performing a combined sequence- and structure-based analysis we detect Asp boxes in more than nine protein families, including bacterial ribonucleases, sulfite oxidases, reelin, netrins, some lipoprotein receptors, and a variety of glycosyl hydrolases. Although the function common to each of these proteins, if any, remains unclear, we discuss possible functions of Asp boxes on the basis of previously determined experimental results and discuss different evolutionary scenarios for the origin of Asp-box containing proteins.
Keywords: Protein evolution, protein structure similarity, protein function, sialidase, reelin, BNR motifs
Detection of sequence, structure, or functional similarities between proteins is central to resolving questions of homology (descent from a common ancestor). Gene products with significant sequence similarity usually have similar structures and functions, and are generally assumed to share a common ancestor (Dayhoff 1976). Other homologs show similarities in their structures (and sometimes functions) even when significant sequence similarities are not detectable (Holm and Sander 1996). In such instances, homology may be inferred from shared derived features, such as key functional residues or unusual structural features (Murzin 1998; Grishin 1999).
A different phenomenon has also been described, where local sequence and structural similarities are apparent in proteins with structures that are otherwise quite different. For example, P-loops, or Walker A motifs, (Gay and Walker 1983) are known to occur in many double wound α/β ATPase structures, for example, Ras p21, and in phosphoenolpyruvate carboxykinase (Matte et al. 1996), which adopts a different α/β fold. A similar situation is observed for the helix-hairpin-helix motif, HhH, (Doherty et al. 1996), which occurs within a variety of nucleic acid-binding domains adopting otherwise different structures. In both instances the motifs share a common molecular function, but the evolutionary explanation for the phenomenon is unclear. One possibility is that non-homologous proteins with different folds have converged on similar sequences; another is that a short ancestral protein was gradually embellished in different ways, leading to different folds with only the original region in common. Lastly, it is possible that these motifs arose from ancient gene duplication and insertion into a variety of different non-homologous proteins.
Russell (1998) recently detected another example of a linear motif that is conserved in different three-dimensional protein folds. The Asp box, a β hairpin, previously identified as a recurring motif in bacterial sialidases (Roggentin et al. 1989; Crennell et al. 1993), was shown to have significant structural similarity to a single hairpin in the immunoglobulin-like (Ig-like)-domain of chitobiase. A total of 17 atoms from identical side chains in the two structures can be superimposed with a RMSD of 0.33 Å with the associated probability of occurrence by chance (P) estimated as 10−23 (Russell 1998). The reason for the occurrence of this motif in non-homologous contexts is unknown, but it may be associated with the known O-glycosyl hydrolase activities of sialidases and chitobiases (Russell 1998).
The method used to identify the chitobiase/sialidase motif detected examples of convergently evolved similarities in molecular function by searching for recurring three-dimensional protein side chain patterns in non-homologous protein folds (Russell 1998). Protein main-chain atoms were ignored, and no requirement was made that the residues of conserved three-dimensional motifs be colinear along polypeptide chains, or that the motifs be restricted to short stretches of sequence. In other words, no additional significance was assigned to the main-chain and sequence order similarity apparent in the chitobiase/neuraminidase motif. By using these additional constraints we have been able to enquire whether more Asp box motifs could be detected with significance from structure and sequence databases, and whether these could provide likely structural, functional, or evolutionary explanations for their occurrence.
Results
Searching for Asp box motifs in known structures
Using the conformation of the Cα atoms from the Asp box-like structure in chitobiase (PDB code 1qba) as a probe, we performed a structural search against species representatives of all protein families in the SCOP database (Lo Conte et al. 2000). The approach is similar to that used previously by Swindells (1993) to identify phosphate-binding loops common to different protein folds, and by Russell and Jackson (2000) to identify putative serine protease inhibitor loops. It is important to emphasize that the technique is a sequence-independent method of searching that is complementary to the sequence-dependent, side-chain-only, structural search approach originally used to identify this motif (Russell 1998).
As expected, this search revealed that the chitobiase β hairpin structure is significantly (P3D < 10−5) similar to sialidase Asp box structures (Table 1). However, very similar structures were also found in an Ig-like fold in sulfite oxidase and in three representatives from a family of microbial ribonucleases that includes barnase and binase (Table 1). The hairpins, and their topological locations, are illustrated in Figure 1 ▶a,b. The putative Asp boxes in chitobiase and sulfite oxidase are both present in a bridge between the two β-sheets of an Ig-like fold. Although the SCOP classification considers these domains as homologs within the E-set family of Ig-like folds, sequence similarity over the domain is not detectable, and no corresponding Asp box-like region is found within other members of either the E-set family or the Ig superfamily. Similarly, only the bacterial, and not the fungal, representatives of the microbial ribonuclease SCOP superfamily (Sevcik et al. 1990) appear to contain significant matches to the Asp box motif.
Table 1.
Asp box motifs detected by rigid body searching
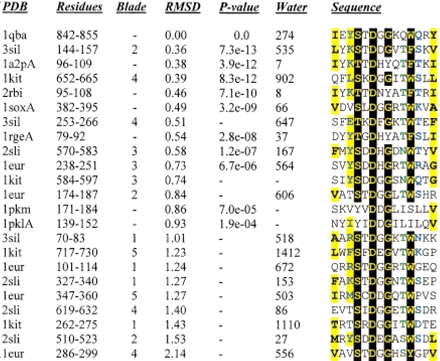 |
The sequences are colored according to Figure 2 ▶. The identifying number of the water molecule associated with each hairpin is given. Two pyruvate kinase structures were detected at values just lower than the 3 × 10−4P-value threshold, and they are not considered by this analysis as containing bona fide Asp boxes due to the absence of a water molecule at positions equivalent to those seen for the sialidases, chitobiase, and ribonucleases (see text for details).
Key to PDB identifiers: 3sil Salmonella typhimurium sialdase; 1a2p Barnase, B. amyloliquefaciens ribonuclease; 1kit Vibrio choerae neuraminidase; 2rbi Mutated binase, B. intermedius ribonuclease; 1sox Gallus gallus sulfite oxidase; 1rge Streptomyces aureofaciens ribonuclease; 2sli Leech trans-sialidase; 1eur Micromonospora viridifaciens sialidase; 1pkm Feline pyruvate kinase; 1pkl Leishmania pyruvate kinase. 1sox & 1qba adopt IG-like folds. 1a2p, 1rge and 2rbi adopt microbial ribonuclease folds. The remaining folds are 6 bladed β propellers. The internal repeat corresponding to each motif is given in the "blade" column for these folds.
Fig. 1.
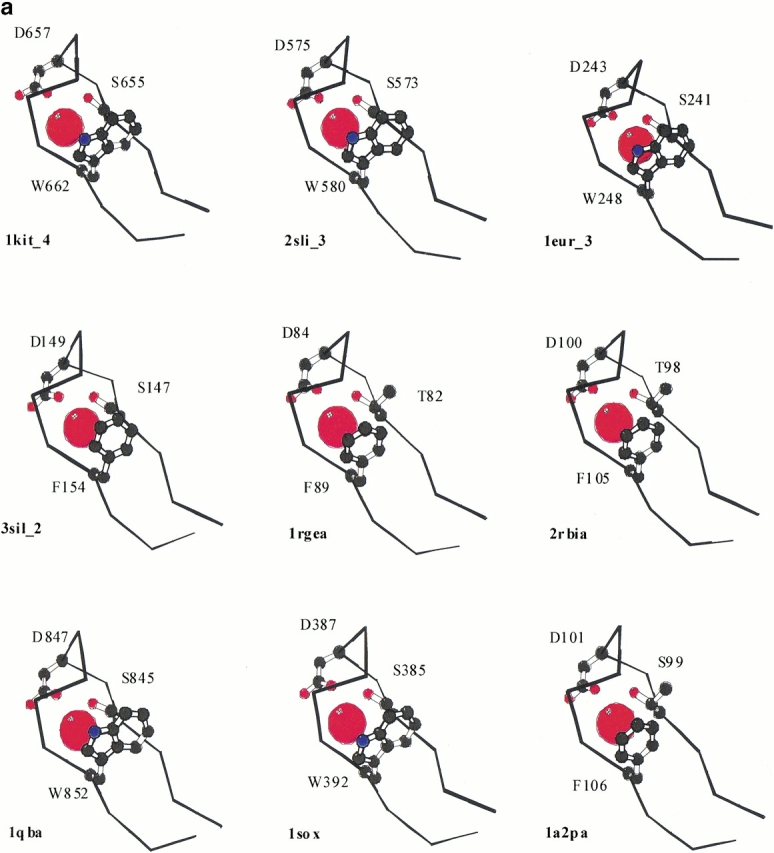

(a) Cα traces of Asp box structures shown in Table 1 represented using Molscript (Kraulis 1991). Only the best match from each of the β propellers is shown. PDB codes are as for Table 1. The side chain atoms of the core conserved residues are shown in ball and stick representation. The one letter amino acid codes and residue numbers are given. Water molecules found in equivalent locations in all structures are illustrated as red spheres. (b) Schematic representation of the location of Asp box motifs within different protein topologies. Amino and carboxy termini are labeled N and C, respectively. Arrows represent β strands, and cylinders α helices. Asp boxes identified in the structural search are boxed with dotted lines.
Although the structures in Table 1 were detected without any consideration of sequence, all share characteristic patterns of residue conservation. For example, three Asp box structures in Serratia marcescens chitobiase, Salmonella typhimurium sialidase, and chicken sulfite oxidase, found using a sequence-independent approach, are significantly similar in sequence; PMACAW = 3.8 × 10−5; as assessed by MACAW (Schuler et al. 1991) using a search space equal to the product of the total lengths of the sequences.
For 18 of the 21 motifs, the central two residues of the matched region reside in the left handed helical region of φ/ψ space, characterizing a type I` β-turn (Lewis et al. 1973). Of the remaining three motifs, two (1kit, blades 1 and 3) show only small deviations from φ/ψ angles favored by type I' turns, whereas the third (1eur, blade 4) represents the most structurally dissimilar motif found. The region of close structural similarity extends well beyond the turn to the whole of a β hairpin (14 residues). Structurally similar turns of all types typically show conservation of two or three consecutive residues central to the turn itself, often glycine and proline residues, that enable or constrain the protein backbone to adopt unusual conformations. To assess the likelihood that the similarity in structure is due solely to the sequence determinants of the type I` turn, we performed similar searches with β hairpin structures, of equal length and containing a central type I` turn. In no cases were significant similarities and residue conservation between different protein folds found (data not shown). The Asp box clearly differs from other turns in being a well conserved structure with sequence conservation apparent for a dozen structurally equivalent positions.
Structurally-equivalent water molecules bound to Asp Boxes
With the exception of one sialidase repeat, each of the identified structures contains a water molecule in a structurally-equivalent position (Fig. 1a ▶ and Table 1). The central aspartate residue of the motif points into the turn, and lies above the conserved water molecule. Well conserved serine or threonine residues at two positions in the motif (Fig. 2 ▶) are likely to coordinate the water molecule. This water appears to be an integral part of the structure, forming hydrogen bonds between the β strands (Fig. 1a ▶). Its appearance in the microbial ribonucleases has been previously noted (Loris et al. 1999). Given the structural and sequence similarity of the microbial ribonuclease Asp box motif to those in sialidases and Ig like folds, it is perhaps not surprising that in hydrated protein crystals, these too should bind water. The detailed similarity does, however, serve to highlight the structural equivalence of the motifs as a whole.
Fig. 2.

Multiple alignment of Asp box sequences. Only one of each family of Asp box sequence-containing proteins has been represented. The majority of Asp box sequences are 14 amino acids in length. However, relative to these, a single reelin Asp box contains a single amino acid insertion, and several other sequences contain a single amino acid deletion. This alignment has been colored using CHROMA (Leo Goodstadt and Chris P. Ponting, unpubl.) and an 80% consensus: Hydrophobic (`h'; ACFGHILMTVWY) residues are highlighted in yellow, conserved residues (>80%) are shown as yellow on black (S and T are treated as equivalent, as are F, W, and Y), big (`b'; EFIKLMQRWY) residues are blue on yellow, small (`s'; ACDGNPSTV) residues are in green, and polar (`p'; CDEHKNQRST) residues are in blue. The sequences shown are: CHB_SERMA, Serratia marcescens chitobiase (GenBank identifier [gi] 3023484); NANH_BACFR, Bacteroides fragilis sialidase (gi 400354); human reelin (gi 4760438); PEP1_YEAST, S. cerevisiae Vps10p (gi 417462); Salmonella typhimurium spi4K (gi 3323596); Avic_ASPAC, Aspergillus aculeatus Avicelase III (gi 3242655); UNC6_CAEEL, C. elegans Unc-6 (gi 465001); H136_ARATH, Arabidopsis thaliana photosystem II stability/assembly factor HCF136 (gi 6016183); FRUA_STRMU, Streptococcus mutans fructanase (gi 2500931); slr1403/SYNY3, Synechocystis sp. slr1403 (gi 1652714); bacteriophage #D endo-N-acetylneuraminidase (gi 3551474); ORF_MYXXA, Myxococcus xanthus ORF (gi 5690376); Ngluc_ENTSP, Enterobacter sp. N-acetyl-beta-D-glucosaminidase (gi 4204206); vrlC/DICNO, Dichelobacter nodosus vrlC (gi 3482864); SLRep/THETH, Thermus thermophilus S-layer repressor (gi 2104901); YkuO/BACSU, Bacillus subtilis YkuO (gi 2632236); APE1882_AERPE, Aeropyrum pernix APE1882 (gi 5105574); and, CSPr_LACDE, Lactobacillus delbrueckii subsp. bulgaricus cell surface proteinase (gi 2127379).
Additional Asp box sequences in sequence databases
A multiple alignment of Asp box sequences detected in the structure database searches was compared with sequence databases using MoST (Tatusov et al. 1994) and HMMer2 (Eddy et al. 1995). Eight protein families were found to contain sequences significantly similar to known Asp boxes (EMoST < 0.05). For each family of Asp box-containing sequences, including those found in the structure database search, the Asp box regions represent among the most highly conserved sequence elements (data not shown). This suggests that these regions are of functional importance. To our knowledge, with the exclusion of sialidase homologs, Asp boxes have been detected previously only in Vps10p (Jorgensen et al. 1999) and in VrlC (Billington et al. 1999). The Asp box sequences detected in a bacteriophage K1F neuraminidase demonstrate the effectiveness of the sequence search strategy, since this protein was not hitherto known to be a homolog of non-phage neuraminidases (Petter and Wimr 1993).
Discussion
Three of these families of Asp box-containing proteins are of particular interest: (1) reelins (D'Arcangelo et al. 1995; D'Arcangelo and Curran 1998), (2) Unc-6/netrin-like axon guidance molecules (Ishii et al. 1991; Serafini et al. 1994; Kennedy et al. 1994) within their laminin N-terminal domains, and (3) LR11–, Vps10p- and sortilin-like neurotensin or sorting receptors (Petersen et al. 1997; Mazella et al. 1998). Curiously, these three families are known to have similar physiological roles in the mammalian central nervous system (CNS). Reelin regulates the migration of cortical plate neurons, Unc-6/netrins represent major cues for neuronal and axonal migration in the embryonic CNS, and LR11/Vps10p/sortilin molecules are members of the lipoprotein receptor family, of which the very-low-density lipoprotein receptor (VLDLR) and apolipoprotein receptor 2 (ApoER2) are known receptors for reelin (Hiesberger et al. 1999; D'Arcangelo et al. 1999).
Functions of Asp box motifs
There is little experimental evidence providing clues to what the common functions mediated by Asp boxes might be. It may be significant that Asp box motifs reside mostly in secreted proteins, with the exception of cytosolic sialidases and sulfite oxidases. It is also worth noting that they occur frequently in proteins that act on, or interact with, polysaccharides. Polysaccharides are often substrates of Asp box-containing glycosyl hydrolases like sialidases, Avicelase III-like endoglucanases, levanases, sucrases, and fructanases, (Russell 1998) and are known to regulate the activities of certain extracellular signalling proteins, such as FGF, when in the form of protein-associated glycosaminoglycans (GAGs). For the latter cases, experimental evidence suggests that GAG is a cofactor for the binding of netrin-1 to its receptor (Bennett et al. 1997). It is thus possible that Asp boxes bind polysaccharides.
A polysaccharide-binding function is unlikely to be found for all Asp box-containing proteins. In barnase-like ribonucleases, for example, the Asp box lies in the active site motif, with the conserved His and Tyr residues interacting with a phosphate group of the bound guanosine 3′-monophosphate ligand. The histidine is essential for catalytic activity and is conserved in all ribonucleases (Sevcik et al. 1990), but is not universally found in the Asp boxes of other proteins.
Evolution of Asp box motifs
The apparently non-homologous proteins in which Asp boxes are found raise questions as to how such a motif evolved. We consider these three possible scenarios: (1) that the sequences and structures have converged in these different folds, (2) that the motifs represent a mobile element that has been duplicated and inserted into different folds, or (3) that the motif is ancestral, representing an ancient component about which structures of modern proteins have been built.
A strong argument for divergence can be made for Asp boxes in sialidases. The presence of multiple Asp boxes within their repeating β sheet structures is explained parsimoniously by gene duplication and divergence of a common ancestral β-propeller blade. Assuming that these motifs have evolved divergently, the level of conservation between repeating copies clearly suggests that these sialidase motifs are resistant to decay by point mutation, a resistance which could be explained by structural, folding, and/or functional constraints. An essential structural role is hard to reconcile with the fact that all Asp box containing fold families we have identified also contain closely related folds without Asp boxes. Similarly, a role for Asp boxes in folding mechanisms is not well supported by experimental work on barnase (Neira and Fersht 1999a,b).
For the remaining Asp boxes however, the case for divergence is more contentious. Propagation via a mobile genetic element (scenario 2, above) would have been less likely for Asp boxes like those in sulfite oxidase where the β hairpin contributes significantly to the fold's hydrophobic core (Fig. 1b ▶). A scenario where Asp boxes have diverged from an ancestral structure (scenario 3) appears at odds with observations that these structures and sequences are absent in homologs (see above).
The evolution by convergence of Asp boxes in non-homologous contexts remains a possibility, if a function, such as a common ligand type, or a critical structural role can be identified.The alternative mechanism, that of a chance convergence of sequence leading to similar energetically favorable structures, is unlikely given the statistically significant sequence similarity, although it is a possibility that cannot be discounted. This would, however, raise the question of why the selective pressures causing convergence between different folds appear to be stronger than those that limit divergence within the β-propellers. Most previously discussed examples of possible sequence convergence have involved short motifs such as integrin-binding RGD tripeptides (Ruoslahti 1996), haem-binding CxxCH pentapeptides (Mathews 1985; Russell 1998), and other metal-binding sites (Russell 1998). Of longer motifs containing more than one secondary structure, only P-loops and HhH motifs might have arisen convergently (Gay and Walker 1983; Doherty et al. 1996). Helix-turn-helix motifs are proposed to be predominantly monophyletic (Rosinski and Atchley 1999), and the active site motifs of types I and II protein phosphatases could have arisen via divergence and circular permutation rather than via convergence as suggested recently (Fauman et al. 1998).
Examples of adaptive convergence (Doolittle 1994) suggest that functional necessity can impose severe constraints on local structure. In cases such as catalytic triad containing serine proteases, sequence order need not be constrained. Even so, Asp boxes may have arisen due to particularly extreme restrictions on the number of structure/sequence solutions that confer a molecular function. If a function for Asp boxes can be established, the possibility that these motifs arose via convergent evolution may be investigated experimentally using in vitro directed evolution approaches (Arnold and Volkov 1999).
Conclusions
The Asp box, HhH, and P-loop motifs represent sequence and structure motifs that are known in different structural contexts. We are currently unable to determine whether these motifs have arisen through divergent and/or convergent evolution. If divergent, then these examples demonstrate that the modular construction of proteins has occurred through the duplication, and insertion elsewhere, of genes representing motifs as well as those representing domains (Doolittle 1995); or they may provide examples of a common peptide ancestor for proteins with otherwise different folds. If convergent, then these three motifs represent some of the first known examples of sequence convergence, as opposed to structure convergence (Doolittle 1994).
Materials and methods
Structure search
Cα coordinates of residues 842–855 of 1qba were used to search against a representative of all structures in SCOP at the species level (Lo Conte et al. 2000). Rigid body fits were performed between the probe structure and all ungapped colinear database fragments of the same length. For example, for a database protein structure of N residues, N-14 fits were performed, by sliding the start position of the probe over the known structure from residue 1 to residue N-14. The RMSD values of the best hit to each protein in the database search were fitted to an extreme value distribution using the `extreme' program of Richard Mott (pers. comm.). This approach allowed the estimation of probabilities, P3D that these RMSD values were detected simply by chance. The lowest P3D-value expected in this search of 3761 structures is of the order 1/3761 = 3 × 10–4. Proteins containing significant matches (P3D < 10–5) to the probe were analyzed for the presence of additional motifs. These additional hits were not subjected to an RMSD threshold, but the presence of the [ST]xDx[GY]xx[WFY] signature (the consensus sequence for the significant hits) was required. The P3D-values generated by this procedure represent the significance of the structural similarity to the original probe, irrespective of evolutionary mechanism (convergence or divergence), and should not be confused with significant results obtained via sequence database searching, which implicitly model the probability of relatedness by divergent evolution.
Sequence search
We compared a multiple alignment block of 21 Asp box sequences (17 from sialidases/neuraminidases, three from bacterial ribonucleases, and one from chitobiase; all pairs <80% identical) with current sequence databases using MoST (Tatusov et al. 1994). On convergence after seven iterations (all pairs <80% identical; EMoST < 0.05), the sequences of eight domain or protein families of unknown tertiary structure were found to be significantly similar to known Asp boxes (EMoST < 0.05). A Hidden Markov model of this alignment and HMMer2 (Eddy et al. 1995) were used to detect additional outlier repeats in these proteins. A subset of these sequences is detected by Pfam (Bateman et al. 2000) (family BNR).
Sequences of structures predicted to contain Asp box motifs were analyzed using MACAW (Schuler et al. 1991), and P-values (PMACAW) were estimated using a searchspace equal to the product of the sequences' lengths. Sequence families (Table 2) were derived using the grouper command of SEALS (Walker and Koonin 1997) and a bits score threshold of 50.
Table 2.
Families of Asp boxes containing proteins detected from searches of protein sequence databases
| Family | Name | Max. no. repeats | Phyletic distribution |
| 1 | Sialidases, neuraminidases | 5 | Bacteria, Eukarya |
| 2 | Reelin | 16 | Eukarya |
| 3 | Vps10p/Pep1p, sortilin, LR11 | 13 | Eukarya |
| 4 | 2 Salmonella ORFs | 4 | Bacteria |
| 5 | Avicelase III homologs | 7 | Bacteria |
| 6 | Unc-6, netrins, laminin N-terminal domains | 1 | Eukarya |
| 7 | YCF48, HCF136 | 4 | Bacteria, Eukarya |
| 8 | Endo-levanases, sucrase, fructanase | 2 | Bacteria |
| Others | slr1403, phage neuraminidase, unknown ORFs, vrlC, S-layer repressor, cell surface proteinase | Various | Archaea, Bacteria, Eukarya |
Acknowledgments
We thank Peer Bork, Martijn Huynen, other Bork group members at EMBL, Jim Fickett, David Searls, and Andrei Lupas for encouragement and helpful discussions. We thank anonymous referees for constructive comments.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertizement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.31901
References
- Arnold, F.H. and Volkov, A.A. 1999. Directed evolution of biocatalysts. Curr. Opin. Chem. Biol. 3 54–59. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Howe, K.L.., and Sonnhammer, E.L.L. 2000. The Pfam protein family database. Nucl. Acids Res. 28 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett K.L., Bradshaw J., Youngman T., Rodgers J., Greenfield B., Aruffo A., and Linsley, P.S. 1997. Deleted in colorectal carcinoma (DCC) binds heparin via its fifth fibronectin type III domain. J. Biol. Chem. 272 26940–26946. [DOI] [PubMed] [Google Scholar]
- Billington, S.J., Huggins, A.S., Johanesen, P.A., Crellin, P.K., Cheung, J.K., Katz, M.E., Wright, C.L., Haring, V., and Rood, J.I. 1999. Complete nucleotide sequence of the 27-kilobase virulence related locus (vrl) of Dichelobacter nodosus: Evidence for extrachromosomal origin. Infect. Immun. 67 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crennell, S.J., Garman, E.F., Laver, W.G., Vimr, E.R., and Taylor, G.L. 1993. Crystal structure of a bacterial sialidase (from Salmonella typhimurium LT2) shows the same fold as an influenza virus neuraminidase. Proc. Natl. Acad. Sci. USA 90 9852–9856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo, G., Miao, G.G., Chen, S.C., Soares, H.D., Morgan, J.I., and Curran, T. 1995. A protein related to extracellular matrix proteins deleted in mouse mutant reelin. Nature 374 719–723. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo, G., and Curran, T. 1998. Reeler: new tales of an old mutant mouse. Bioessays 20 235–244. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D.S., Sheldon, M., and Curran, T. 1999. Reelin is a ligand for lipoprotein receptors. Neuron 24 471–479. [DOI] [PubMed] [Google Scholar]
- Dayhoff, M.O. 1976. The origin and evolution of protein superfamilies. Fed. Proc. 35 2132–2138. [PubMed] [Google Scholar]
- Doherty, A.J., Serpell, L.C.,and Ponting, C.P. 1996. The helix-hairpin-helix DNA-binding motif: a structural basis for non-sequence- specific recognition of DNA. Nucl Acids Res 24: 2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle, R.F. 1994. Convergent evolution: The need to be explicit. Trends Biochem. Sci. 19 15–18. [DOI] [PubMed] [Google Scholar]
- Doolittle, R.F. 1995. The multiplicity of domains in proteins. Annu. Rev. Biochem. 64 287–314. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R., Mitchison, G., and Durbin, R. 1995. Maximum discriminating hidden Markov models of sequence consensus. J. Comput. Biol. 2 9–23. [DOI] [PubMed] [Google Scholar]
- Fauman, E.B., Cogswell, J.P., Lovejoy, B., Rocque, W.J., Holmes, W., Montana, V.G., Piwnica-Worms, H., Rink, M.J., and Saper, M.A.. 1998. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 93 617–625. [DOI] [PubMed] [Google Scholar]
- Gay, N.J. and Walker, J.E. 1983. Homology between human bladder cancinoma oncogene product and mitochondrial ATP-synthase. Nature 301 262–264. [DOI] [PubMed] [Google Scholar]
- Grishin, N.V. 1999. Phosphatidylinositol phosphate kinase: A link between protein kinase and glutathione synthase folds. J. Mol. Biol. 291 239–247 [DOI] [PubMed] [Google Scholar]
- Hiesberger, T., Trommsdorff, M., Howell, B.W., Goffinet, A., Mumby, M.C., Cooper, J.A., and Herz, J. 1999. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24 481–489. [DOI] [PubMed] [Google Scholar]
- Holm, L and Sander, C. 1996. Mapping the protein universe. Science 273 595–603. [DOI] [PubMed] [Google Scholar]
- Ishii, N., Wadsworth, W.G., Stern, B.D., Culotti, J.G., and Hedgecock, E.M. 1992. UNC-6, a laminin-related protein, guides cell and pioneer axon migrations in C. elegans. Neuron 9 873–881. [DOI] [PubMed] [Google Scholar]
- Jackson, R.M. and Russell, R.B. 2000. The serine protease inhibitor canonical loop conformation: examples found in extracellular hydrolases, toxins, cytokines and viral proteins. J. Mol. Biol. 296 325–334. [DOI] [PubMed] [Google Scholar]
- Jorgensen, M.U., Emr, S.D., and Winther, J.R. 1999. Ligand recognition and domain structure of Vps10p, a vacuolar protein sorting receptor in Saccharomyces cerevisiae. Eur. J. Biochem. 260 461–469. [DOI] [PubMed] [Google Scholar]
- Kennedy, T.E., Serafini, T., de la Torre, J.R., and Tessier-Lavigne, M. 1994. Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell 78 425–435. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 24: 946–950 [Google Scholar]
- Lewis, P.N., Momany, F.A., and Scheraga, H.A. 1973. Chain reversals in proteins. Biochim. Biophys. Acta. 303 211–229. [DOI] [PubMed] [Google Scholar]
- Lo Conte, L., Ailey, B., Hubbard, T.J., Brenner, S.E., Murzin, A.G., and Chothia, C. 2000. SCOP: A structural classification of proteins database. Nucl. Acids Res. 28 257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loris, R., Langhorst, U., De Vos, S., Decanniere, K., Bouckaert, J., Maes, D., Transue, T.R., and Steyaert, J. 1999. Conserved water molecules in a large family of microbial ribonucleases. Proteins 36 117–134 [DOI] [PubMed] [Google Scholar]
- Mathews, FS. 1985. The structure, function and evolution of cytochromes. Prog. Biophys. Mol. Biol. 45 1–56. [DOI] [PubMed] [Google Scholar]
- Matte, A., Goldie, H., Sweet, R.M., and Delbaere, L.T. 1996. Crystal structure of Escherichia coli phosphoenolpyruvate carboxykinase: A new structural family with the P-loop nucleoside triphosphate hydrolase fold. J. Mol. Biol. 256 126–143. [DOI] [PubMed] [Google Scholar]
- Mazella, J., Zsurger, N., Navarro, V., Chabry, J., Kaghad, M., Caput, D., Ferrara, P., Vita., N., Gully, D., Maffrand, J.P., and Vincent, J.P. 1998. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. .J Biol. Chem. 273 26273–26276. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G. 1998. How far divergent evolution goes in proteins. Curr. Opin. Struct. Biol. 8 380–387 [DOI] [PubMed] [Google Scholar]
- Neira J.L. and Fersht, A.R. 1999a. Exploring the folding funnel of a polypeptide chain by biophysical studies on protein fragments. J. Mol. Biol. 285 1309–1333 [DOI] [PubMed] [Google Scholar]
- Neira, J.L. and Fersht, A.R. 1999b. Acquisition of native-like interactions in C-terminal fragments of barnase. J. Mol. Biol. 287 421–432 [DOI] [PubMed] [Google Scholar]
- Petersen, C.M., Nielsen, M.S., Nykjaer, A., Jacobsen, L., Tommerup, N., Rasmussen, H.H., Roigaard, H., Gliemann, J., Madsen, P., and Moestrup, S.K. 1997. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J. Biol. Chem. 272 3599–3605. [DOI] [PubMed] [Google Scholar]
- Petter, J.G. and Vimr, E.R. 1993. Complete nucleotide sequence of the Bacteriophage K1F tail gene encoding Endo-N-acylneuraminidase (Endo-N) and comparison to an Endo-N homolog in Bacteriophage PK1E. J. Bacteriol. 175 4354–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggentin, P., Rothe, B., Kaper, J.B., Galen, J., Lawrisuk, L., Vimr, E.R., and Schauer, R. 1989. Conserved sequences in bacterial and viral sialidases. Glycoconjugate J. 6 349–353. [DOI] [PubMed] [Google Scholar]
- Rosinski, J.A. and Atchley WR. 1999. Molecular evolution of helix-turn-helix proteins. J. Mol. Evol. 49 301–309. [DOI] [PubMed] [Google Scholar]
- Ruoslahti, E. 1996. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12 697–715. [DOI] [PubMed] [Google Scholar]
- Russell, RB. 1998. Detection of protein three-dimensional side-chain patterns: new examples of convergent evolution. J. Mol. Biol. 279 1211–1227. [DOI] [PubMed] [Google Scholar]
- Schuler, G.D., Altschul, S.F., and Lipman, D.J. 1991. A workbench for multiple alignment construction and analysis. Proteins 9 180–190. [DOI] [PubMed] [Google Scholar]
- Serafini, T., Kennedy, T.E., Galko, M.J., Mirzayan, C., Jessell, T.M., Tessier-Lavigne, M. 1994. The netrins define a family of axon outgrowth-promoting protenis homologous to C. elegans UNC-6. Cell 78 409–424. [DOI] [PubMed] [Google Scholar]
- Sevcik, J., Sanishvili, R.G., Pavlovsky, A.G., Polyakov, K.M. 1990. Comparison of active sites of some microbial ribonucleases: structural basis for guanylic specificity. Trends Biochem. Sci. 15 158–162. [DOI] [PubMed] [Google Scholar]
- Swindells, M.B. 1993. Classification of doubly wound nuceotide binding topologies using automated loop searches. Protein Sci. 2 2146–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Altschul, S.F., and Koonin, E.V. 1994. Detection of conserved segments in proteins: Iterative scanning of sequence databases with alignment blocks. Proc. Natl. Acad. Sci. USA 91 12091–12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, D.R. and Koonin, E.V. 1997. SEALS: A system for easy analysis of lots of sequences. Intelligent Systems for Molecular Biology 5 333–339. [PubMed] [Google Scholar]