

Abstract
We have examined the irreversible inactivation mechanism of the membrane protein diacylglycerol kinase in the detergents n-octyl-β-D-glucopyranoside (OG) at 55°C and n-decyl-maltopyranoside (DM) at 80°C. Under no inactivation conditions did we find any direct evidence for the chemical modifications that are commonly found in soluble proteins. Moreover, protein inactivated at 55°C in OG could be reactivated by an unfolding and refolding protocol, suggesting that the protein is inactivated by a stable conformational change, not a covalent modification. We also found that the inactivation rate decreased with both increasing protein concentration and increasing thermodynamic stability, consistent with an inactivation pathway involving transient dissociation and/or unfolding of the protein. Our results suggest that the primary cause of diacylglycerol kinase inactivation is not low solubility, but poor intrinsic stability in the detergent environment.
Keywords: Stability, irreversible inactivation, denaturation, aggregation, half life
Whereas membrane proteins can be extremely stable in the bilayer (Haltia and Freire 1995; White and Wimley 1998; White and Wimley 1999), many membrane proteins are poorly behaved in detergent solution and undergo rapid aggregation or inactivation. The resulting technical problems can make biochemical, biophysical, and structural characterization difficult if not impossible. It would therefore be advantageous to develop methods that yield better kinetic stability of membrane proteins. To achieve this goal, however, it might be necessary to expand our understanding of why membrane proteins are kinetically unstable in detergent solution.
Although the primary pathways for membrane protein inactivation in detergent are currently unknown, irreversible inactivation of soluble proteins has been well studied (Cowan 1995; Cowan 1997; Daniel et al. 1996; Klibanov 1983; Mozhaev 1993; Mozhaev et al. 1988). The causes of soluble protein inactivation can be divided into two groups: covalent modification or conformational change. Examples of the covalent alterations that have been observed include side-chain oxidation, destruction of disulfide bonds, formation of incorrect disulfide bonds, deamidation, racemization, and hydrolysis of peptide bonds. The noncovalent changes include aggregation as well as the formation of incorrect and kinetically trapped conformations (Ahern and Klibanov 1985; Mulkerrin and Wetzel 1989; Nury and Meunier 1990; Tomazic and Klibanov 1988; Tomizawa, et al. 1994; Zale and Klibanov 1986).
In soluble proteins, the pathway to covalently and conformationally inactivated states generally passes through the unfolded state of the protein (Klibanov 1983; Lumry and Eyring 1954; Mozhaev 1993). These results can be understood easily because the unfolded state of the protein exposes hydrophobic surfaces that could lead to stable conformational rearrangements or aggregation. The unfolded state is also more flexible and consequently more amenable to conformations that favor chemical modification.
Here we examined the irreversible inactivation of a specific integral membrane protein, diacylglycerol kinase (DGK), from Escherichia coli. DGK is a 121 residue, trimeric protein, which catalyzes the conversion of diacylglycerol to phosphatidic acid. The predicted topology of DGK consists of two cytoplasmic helices and three transmembrane helices (Lightner et al. 1983; Loomis et al. 1985; Smith et al. 1994). We investigated both the nature of the inactive state and the pathway to the inactive state.
Results
Conditions for inactivation
Inactivation mechanisms can vary with the solvent system used. In this study we examined inactivation mechanisms using two different detergents, OG and DM, under buffer conditions that are not unusual for biochemical experiments: 50 mM sodium phosphate (pH 7.5) and 0.3 M NaCl. The protein is much more resistant to thermal inactivation in DM than in OG so it was necessary to use different temperatures for the two detergents. For OG, a temperature of 55°C was used and for DM, the inactivation temperature was 80°C. At 0.3 mg/ml, the half-life of DGK in OG at 55°C is 6 min, and in DM at 80°C, the half-life is 35 min.
Covalent modification was not detected
To test whether covalent modification plays an important role in DGK inactivation, we carried out electrospray mass spectroscopy of native DGK and heat-inactivated DGKs. As shown in Table 1, no significant change in molecular weight was observed for DGK inactivated in OG at 55°C or DM at 80°C. There were also no significant changes in the size and intensity of DGK bands after sds-page (data not shown). These results rule out most common covalent modifications that significantly alter molecular weight, such as the formation of new intersubunit disulfide bonds, peptide bond hydrolysis, or oxidation of amino acid residues.
Table 1.
Electrospray mass spectroscopy of unheated and heat-treated DGK
| Activity remaining (%) | Mass (Da) | |
| Unheated | 100 | 14236.1 ± 1.1 |
| 55°C in 1.5% OG, 50 min | 40 | 14236.7 ± 1.2 |
| 80°C in 0.5% DM, 60 min | 28 | 14235.6 ± 1.4 |
Some covalent modifications, such as intramolecular disulfide bond formation, deamidation, and racemization, which have been found to be responsible for inactivation of some soluble proteins, cannot be detected by our mass spectroscopy experiment. Additional experiments were therefore conducted to test for covalent alterations resulting in small mass changes.
Asn and Gln residues are susceptible to deamidation. Deamidation would result in a molecular weight change of about 1 dalton, which is within the experimental error of mass spectroscopy.
Because DGK contains four Asn and one Gln, we tested whether deamidation occurs during irreversible inactivation. Deamidation was tested by assaying the release of NH4+ using the method of Nazar and Schoolwerth (1979). As shown in Figure 1 ▶, no significant deamidation was detected when DGK was inactivated in OG at 55°C or DM at 80°C. Under our conditions, the assay could detect less than 30% deamidation of only one amino acid residue. Thus, the loss of activity cannot be accounted for by deamidation.
Fig. 1.

Deamidation during irreversible inactivation. DGK (2–3 mg/ml) was heated for the indicated time periods at 55°C in 50 mM sodium phosphate (pH 7.5), 0.3 M NaCl containing 1.5% OG, or at 80°C in the same buffer containing 0.5% DM. Filled circles: activity of sample in OG. Filled triangles: activity of sample in DM. Open circles: number of Asp/Asn residues in OG sample deamidated. Open triangles: number of Asp/Asn residues in DM sample deamidated.
Racemization of amino acids is another chemical modification that cannot be detected by mass spectroscopy because it is not accompanied by a mass change. We assayed for racemization at the susceptible amino acids, Asp and Asn. The amount of D-Asx was measured before and after heat inactivation using the method of Aswad (1984). In this method, the protein is hydrolyzed and the free amino acids derivatized with N-acetyl-L-cysteine (NAC). Derivatized D-Asp and L-Asp can then be separated by reverse-phase HPLC. Because racemization can also occur during protein hydrolysis, we compared the amount of D-Asp in the same sample before and after heat inactivation. We found only a trace of D-Asp before and after heat inactivation in DM or OG (data not shown). Racemization at Asp or Asn can therefore not account for the loss of activity.
Another possible covalent change that we could not detect by mass spectroscopy is intramolecular disulfide formation. However, no difference in inactivation rate was observed when DGK was inactivated in the presence or absence of 10 mM DTT (data not shown). Moreover, no altered mobility was observed on SDS gels before and after heat inactivation. Thus, intramolecular disulfide bond formation is unlikely to be responsible for DGK inactivation.
Reactivation of DGK
In the results presented so far, we were unable to detect likely chemical modification events. To further test whether the inactivation of DGK results from a covalent or noncovalent process, we attempted to reactivate DGK after inactivation by heat treatment. If inactivation of DGK is the result of conformational changes and not covalent modification, it might be possible to recover activity by first completely unfolding and then refolding the protein. On the other hand, if the inactivation is due to covalent modification, unfolding and refolding will not reverse the inactivation process. It is known that DGK can be refolded after unfolding in SDS (Lau et al. 1999). We therefore attempted to reactivate thermally inactivated DGK by first dissolving the inactive/aggregated protein in SDS and then diluting the protein in a nondenaturing detergent/lipid combination (see Materials and Methods).
As shown in Figure 2A ▶ for DGK inactivated in OG at 55°C, the ability to recover DGK activity does not diminish with increasing inactivation times. This suggests that under the conditions of our inactivation experiment, inactivation is not the result of covalent changes. For DGK inactivated in DM at 80°C, however, we were unable to restore activity after inactivation (Fig. 2B ▶). Thus, we cannot rule out the possibility of some covalent changes occurring under these conditions that we could not detect directly.
Fig. 2.


Reactivation of DGK after inactivation by heat treatment. DGK samples (0.3 mg/ml) were heated (A) at 55°C in 50 mM sodium phosphate (pH 7.5), 0.3 M NaCl containing 1.5% OG, or (B) at 80°C in the same buffer containing 0.5% DM. Each sample was completely denatured and renatured as described in Materials and Methods. The residual activity after heat treatment (•) and after renaturation (○) was normalized to the activities of unheated DGK which underwent the same treatment. The data represent the average and standard deviation of three independent experiments.
We tested whether the different refolding results were due to the change in temperature rather than to the detergent by inactivating the protein in OG at 80°C. Under these conditions, we were similarly unable to recover activity after unfolding and refolding. Thus, it appears that the inactive state at high temperatures is different than the inactive state at lower temperatures.
Because we could not detect any covalent modifications occurring at 80°C, our inability to recover activity could be the result of a different inactive conformation, such that SDS denaturation does not lead to the same refoldable conformation. Because of copious aggregation, it was not possible to probe directly the conformation of the inactive proteins spectroscopically, but it was possible to collect CD spectra of the inactivated proteins after resolubilizing in SDS.
The near-UV CD spectra of SDS-solubilized DGK that had been inactivated in OG at 55°C and DM at 80°C are shown in Figure 3 ▶. The spectra are significantly different. These results indicate that conformational differences could account for the different reactivation behavior of DGK at the two temperatures.
Fig. 3.

CD spectra of inactivated DGK after dissolving in SDS. Solid line: DGK inactivated in OG at 55°C. Dashed line: DGK inactivated in DM at 80°C. The remaining activity in the OG sample was 32%, and in the DM sample it was 25%. Both inactivated samples were solubilized in SDS before recording the spectra (see Materials and Methods).
Concentration dependence of irreversible inactivation
On thermal inactivation of DGK, aggregate is clearly visible. Thus, one might expect that the rate of inactivation would increase with increasing protein concentration. However, we find that the opposite is true. As shown in Figure 4 ▶ in both OG and DM, the rate of inactivation slows with increasing protein concentration. These results suggest that dissociation of the trimeric DGK is an important step on the inactivation pathway (see below).
Fig. 4.


Irreversible inactivation rate of DGK as a function of protein concentration. Inactivation was carried out (A) at 55°C for 10 min in 50 mM sodium phosphate (pH 7.5), 0.3 M NaCl containing 1.5% OG, or (B) at 75°C for 30 min in the same buffer containing 0.5% DM. The residual activities after inactivation were normalized by the activity of unheated enzyme. The data represent the average and standard deviation for three sets of independent experiments.
Correlation of inactivation rate with thermodynamic stability
If the predominant pathway to the inactive state passes through the unfolded state, kinetic stability should be correlated with thermodynamic stability. This was tested by measuring the kinetic and thermodynamic stabilities of a collection of cysteine substitution mutants we constructed earlier (Lau et al. 1999). The thermodynamic stability of DGK can be measured using a chemical denaturation assay (Lau and Bowie 1997) in which the protein is placed in the nondenaturing detergent DM and unfolded by adding increasing concentrations of the denaturing detergent SDS. When denaturation is monitored by changes in absorbance at 294 nm, two reversible unfolding transitions are observed. The transition midpoints can be used as a measure of thermodynamic stability. As shown in Figure 5 ▶, we see a good correlation between half-life and thermodynamic stability.
Fig. 5.

Correlation between kinetic and thermodynamic stability. Half-lives were measured in DM at 1.7 mg/ml where the reaction approximates first-order kinetics. The relative half-life is the half-life of the mutant divided by the half-life of the wild-type protein. [SDS]1/2 is the SDS concentration at the midpoint of the second phase of the SDS denaturation curves and taken as the point of maximum slope. In addition to the wild-type protein, data are shown for cysteine substitution mutants at the following sixteen positions: 46, 47, 53, 54, 55, 56, 57, 59, 60, 61, 62, 63, 64, 66, 68, and 70.
Discussion
The cause of irreversible inactivation can be divided into two groups: noncovalent conformational alterations and covalent changes. Our results suggest that conformational change is the major inactivation mechanism for DGK, particularly at lower temperatures. We tested directly for the covalent modifications that have been found in other proteins (Ahern and Klibanov 1985; Mulkerrin and Wetzel 1989; Tomazic and Klibanov 1988; Tomizawa et al. 1994; Zale and Klibanov 1986) and found no evidence for these changes. Moreover, protein inactivated at 55°C in OG could be reactivated by an unfolding and refolding protocol. These results essentially preclude a covalent change as the proximate cause of inactivation. On the other hand, we were unable to reactivate the enzyme after inactivation at 80°C in either DM or OG. Thus, we cannot rule out inactivation by covalent change at higher temperatures. The fact that we do not directly observe any covalent changes, however, could indicate that a different, more severe conformational change might occur at high temperature that is irreversible in SDS. Consistent with this possibility, spectroscopic evidence suggests that the protein inactivated at 80°C does adopt a different conformation than the protein inactivated at 55°C.
We do not know the exact nature of the conformational change that occurs at low temperature (and possibly at high temperature), but some of the protein ends in visible aggregate. Aggregation and precipitation is quite commonly observed for membrane proteins after extraction into detergent. One possible reason that membrane proteins are prone to aggregation is that detergent molecules are unable to completely mask the hydrophobic surface of the membrane protein. For example, transient detergent dissociation could lead to exposure of a hydrophobic patch, nucleating aggregation. If this were the primary cause of aggregation, however, it might be expected that the inactivation rate would increase with increasing protein concentration.
However, we find the opposite behavior for DGK. In both detergents tested here, DGK is more stable at higher protein concentrations.
The observed concentration dependence can be explained by an inactivation pathway that involves subunit dissociation as follows:
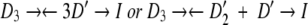 |
1 |
where D3 is the folded, trimeric DGK enzyme, D′2 and D′ are the dissociated, possibly unfolded dimeric and monomeric species, and I represents the inactive form. Additional evidence that dissociation and unfolding precedes inactivation is provided by the good correlation between thermodynamic stability and kinetic stability. Moreover, mutants that improve the kinetic stability of DGK run as stable trimers on SDS gels (Lau et al. 1999; Zhou and Bowie 2000). This mechanism is in agreement with the results from soluble proteins in which the major pathways pass through the unfolded state (Klibanov 1983; Lumry and Eyring 1954; Mozhaev 1993). Although we have examined inactivation in only two detergents, we have found that mutations, which alter the rate of inactivation in OG, also alter inactivation rates in five other detergents (Zhou and Bowie 2000), suggesting that similar inactivation mechanisms operate.
Our results suggest that efforts to improve the behavior of membrane proteins should target thermodynamic stability by altering solvent conditions, by adding compounds that bind the native state (Zhou et al. 1997), or by the introduction of stability-enhancing mutations (Lau et al. 1999; Zhou and Bowie 2000).
Materials and methods
The detergents n-Octyl-β-glucoside (OG) and n-Decyl-β-maltoside (DM) were obtained from Anatrace. Bovine heart cardiolipin and 1,2-Dioleoyl-sn-glycerol (DAG) were obtained from Avanti Polar Lipids, and 1-pyrenebutanoic acid was obtained from Molecular Probes. All other chemicals were from either Fisher or Sigma.
Protein purification and enzyme activity assay
DGK was purified as described previously (Lau and Bowie 1997). Purified enzymes were stored at −80°C in a buffer containing 50 mM sodium phosphate, 0.3 M NaCl, 0.25 M imidazole (pH 8), and 0.5% (w/v) DM. Activities of DGK were measured using a colorimetric assay system (Badola and Sanders 1997; Lau and Bowie 1997; Lau et al. 1999). In this assay, ADP generated by the DGK catalyzed reaction was coupled to the oxidation of NADH, which was then measured by changes in absorbance at 340 nm using a microplate reader (Molecular Devices).
Stability measurements
Irreversible inactivation was carried out as described (Lau et al. 1999; Zhou and Bowie 2000). Briefly, a protein stock was diluted to the desired concentration (standard conditions were 0.3 mg/ml) in 50 mM sodium phosphate (pH 7.5), 0.3M NaCl with either 1.5% (w/v) OG or 0.5% (w/v) DM. Following a 30 min incubation on ice to recover full activity after thawing the stock enzymes, 80 μl aliquots of the diluted samples were heated at the desired temperature in a thermocycler (MJ research), then transferred to ice at given time intervals. The residual activities of each sample were then assayed. Inactivation was quantified by the rate at which activity dropped. Thermodynamic stability was measured using the chemical denaturant assay as described (Lau and Bowie 1997).
Reactivation of DGK inactivated by heat treatment
Reactivation was carried out using a modified SDS unfolding and refolding method (Lau et al. 1999). To unfold, the samples were diluted fivefold in a buffer containing 10 mM Pipes (pH 7) and 1% SDS. To refold, the samples were diluted 10-fold in a buffer containing 50 mM Pipes (pH 7), 50 mM DM, 2 mM bovine heart cardiolipin, and 1.5 mM DTT. After incubation at 4°C overnight, the activity of each sample was assayed. For unheated DGK, 50% enzyme activity can be recovered. Therefore, the data were normalized relative to the activity of the unheated sample which underwent the same unfolding and refolding treatment.
Circular dichroism spectroscopy
DGK was heat inactivated in 50 mM sodium phosphate (pH 7.5) and 300 mM NaCl with either 1.5% OG or 0.5% DM. The sample in OG contained 7.1 mg/ml DGK and was heated at 55 °C until 32% activity remained. The sample in DM contained 6.3 mg/ml DGK and was heated at 80°C until 25% activity remained. Considerable visible aggregate was present in the inactivated samples. The inactivated proteins were diluted fivefold in 10 mM PIPES (pH 7) and 1% SDS which clarified the samples. CD spectra of the resolubilized protein samples were recorded on an AVIV 62DS spectrometer using 1 cm pathlength cells. Circular dichroism was measured at 1 nm intervals. Thirty spectra of both buffer blanks and samples were collected and averaged.
Electrospray mass spectroscopy
To prepare samples for electrospray mass spectroscopy, we used the procedure of Whitelegge and coworkers (1999). Each 100 μl aliquot of 0.6 mg/ml DGK was mixed with 300 μl methanol, 100 μl chloroform, and 200 μl water, and spun in a microfuge for 1 min. The proteins were in the interface after the centrifugation. After removing the upper aqueous phase, 300 μl of methanol was mixed with each sample and centrifuged for 1 min.
The pellets were washed once with 1 ml water and stored at −80°C. Immediately before injection, protein pellets were dissolved in 60%–90% formic acid, then diluted twofold in 50% acetonitrile, 50% H2O and 0.1% HCOOH.
Determination of deamidation
Due to the precipitation of inactivated proteins, we could not use isoelectric focusing to detect deamidation of Asn or Gln. Instead, deamidation was investigated by assaying the amount of ammonia formed during irreversible inactivation. The method, as described by Nazar and Schoolwerth (1979), was an enzymatic assay utilizing the following reaction catalyzed by glutamate dehydrogenase: α-Ketoglutarate + NH4+ + NADH → Glutamate + NAD+ + H2O
The fluorescence of NADH was measured with a FL900CDT Time-Resolved fluorometer (Edinburgh Analytical Instruments). To minimize measurement error, 100–200 pg/ml 1-pyrenebutanoic acid was added as an internal standard. The excitation wavelength was 340 nm, and the emission was detected at 378 nm (E378) for 1-pyrenebutanoic acid and 470 nm (E470) for NADH. The ratio E470 : E378 reflects the relative amount of NADH.
To prevent the release of ammonia in gaseous form, the inactivation reaction was carried out at pH 6.5 or 7.5 in sealed glass ampules.
Acknowledgments
This work was supported by NIH grants RO1GM47485 and RO1GM59164. We thank Frank Pettit, Sarah Yohannan, Salem Faham, Aaron Chamberlain, and David Grosfeld for comments on the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.34201
References
- Ahern, T. and Klibanov, A. 1985. The mechanism of irreversible enzyme inactivation at 100C. Science 228 1280–1284. [DOI] [PubMed] [Google Scholar]
- Aswad, D. 1984. Determination of D- and L-Aspartate in amino acid mixtures by high-performance liquid chromatography after derivatization with a chiral adduct of o-phthaldialdehyde. Anal. Biochem. 137 405–409. [DOI] [PubMed] [Google Scholar]
- Badola, P. and Sanders, C. 1997. E. coli diacylglycerol kinase is an evolutionarily optimized membrane enzyme and catalyzes direct phosphoryl transfer. J. Biol. Chem. 272 24176–24182. [DOI] [PubMed] [Google Scholar]
- Cowan, D. 1995. Protein stability at high temperatures. Essays Biochem. 29 193–207. [PubMed] [Google Scholar]
- Cowan, D. 1997. Thermophilic proteins: Stability and function in aqueous and organic solvents. Comp. Biochem. Physiol. 118A 429–438. [DOI] [PubMed] [Google Scholar]
- Daniel, R., Dines, M., and Petach, H. 1996. The denaturation and degradation of stable enzymes at high temperatures. Biochem. J. 317 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltia, T. and Freire, E. 1995. Forces and factors that contribute to the structural stability of membrane proteins. Biochim. Biophys. Acta 1228 1–27. [DOI] [PubMed] [Google Scholar]
- Klibanov, A. 1983. Stabilization of enzymes against thermal inactivation. Adv. Appl. Microbiol. 29 1–28. [DOI] [PubMed] [Google Scholar]
- Lau, F. and Bowie, J. 1997. A method to assess the stability of a membrane protein. Biochemistry 36 5884–5892. [DOI] [PubMed] [Google Scholar]
- Lau, F., Chen, X., and Bowie, J. 1999. Active sites of diacylglycerol kinase from Escherichia coli are shared between subunits. Biochemistry 38 5521–5527. [DOI] [PubMed] [Google Scholar]
- Lau, F., Nauli, S., Zhou, Y., and Bowie, J. 1999. Changing single side chains can greatly enhance the resistance of a membrane protein to irreversible inactivation. J. Mol. Biol. 290 559–564. [DOI] [PubMed] [Google Scholar]
- Lightner, V., Bell, R., and Modrich, P. 1983. The DNA sequences encoding plsB and dgk loci of Escherichia coli. 258 10856. [PubMed] [Google Scholar]
- Loomis, C., Walsh, J., and Bell, R. 1985. sn-1,2-Diacylglycerol kinase of Escherichia coli. J. Biol. Chem. 260 4091. [PubMed] [Google Scholar]
- Lumry, R. and Eyring, H. 1954. Conformational changes of proteins. J. Phys. Chem. 58 110. [Google Scholar]
- Mozhaev, V. 1993. Mechanism-based strategies for protein thermostabilization. Trends Biotechnol. 11 88–95. [DOI] [PubMed] [Google Scholar]
- Mozhaev, V., Berezin, I., and Martinek, K. 1988. Structure-stability relationship in proteins: Fundamental tasks and strategy for the development of stabilized enzyme catalysts for biotechnology. CRC Critical Reveiws in Biochemistry 23 235–281. [DOI] [PubMed] [Google Scholar]
- Mulkerrin, M. and Wetzel, R. 1989. pH dependence of the reversible and irreversible thermal denaturation of gamma interferons. Biochemistry 28 6556–6561. [DOI] [PubMed] [Google Scholar]
- Nazar, B. and Schoolwerth, A. 1979. An improved microfluorometric enzymatic assay for the determination of ammonia. Anal. Biochem. 95 507–511. [DOI] [PubMed] [Google Scholar]
- Nury, S. and Meunier, J. 1990. Molecular mechanisms of the irreversible thermal denaturation of guinea-pig liver transglutaminase. Biochem. J. 266 487–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, R., O'Toole, J., Maguire, M., and Sanders, C. 1994. Membrane topology of Escherichia coli diacylglycerol kinase. J. Bact. 176 5459–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomazic, S. and Klibanov, A. 1988. Mechanisms of irreversible thermal inactivation of Bacillus alpha-amylases. J. Bio. Chem. 263 3086–3091. [PubMed] [Google Scholar]
- Tomizawa, H., Yamada, H., and Imoto, T. 1994. The mechanism of irreversible inactivation of lysozyme at pH 4 and 100°C. Biochemistry 33 13032–13037. [DOI] [PubMed] [Google Scholar]
- White, S. and Wimley, W. 1998. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta 1376 339–352. [DOI] [PubMed] [Google Scholar]
- White, S. and Wimley, W. 1999. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 28 319–365. [DOI] [PubMed] [Google Scholar]
- Whitelegge, J., le Coutre, J., Lee, J., Engel, C., Prive, G., Faull, K., and Kaback, H. 1999. Toward the bilayer proteome, electrospray ionization-mass spectrometry of large, intact membrane proteins. Proc. Natl. Acad. Sci. USA 96 10695–10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zale, S. and Klibanov, A. 1986. Why does ribonuclease irreversibly inactivate at high temperatures? Biochemistry 25 5432–5444. [DOI] [PubMed] [Google Scholar]
- Zhou, Y. and Bowie, J. 2000. Building a thermostable membrane protein. J. Bio. Chem. 275 6975–6979. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., Wen, J., and Bowie, J. 1997. A passive transmembrane domain. Nature Struc. Biol. 4 986–990. [DOI] [PubMed] [Google Scholar]