

Abstract
Chorionic gonadotropin (hCG) is a heterodimeric placental glycoprotein hormone essential for human reproduction. Twenty hCG β-subunit residues, termed the seatbelt, are wrapped around α-subunit loop 2 (α2) and their positions "latched" by a disulfide formed by cysteines at the end of the seatbelt (Cys 110) and in the β-subunit core (Cys 26). This unique arrangement explains the stability of the heterodimer but raises questions as to how the two subunits combine. The seatbelt is latched in the free β-subunit. If the seatbelt remained latched during the process of subunit combination, formation of the heterodimer would require α2 and its attached oligosaccharide to be threaded through a small β-subunit hole. The subunits are known to combine during oxidizing conditions in vitro, and studies described here tested the idea that this requires transient disruption of the latch disulfide, possibly as a consequence of the thioredoxin activity reported in hCG. We observed that alkylating agents did not modify either cysteine in the latch disulfide (Cys 26 or Cys 110) during heterodimer formation in several oxidizing conditions and had minimal influence on these cysteines during combination in the presence of mild reductants (1–3 mM β-mercaptoethanol). Reducing agents appeared to accelerate subunit combination by disrupting a disulfide (Cys 93–Cys 100) that forms a loop within the seatbelt, thereby increasing the size of the β-subunit hole. We propose a mechanism for hCG assembly in vitro that depends on movements of α2 and the seatbelt and suggest that the process of glycoprotein hormone subunit combination may be useful for studying the movements of loops during protein folding.
Keywords: Glycoprotein hormone subunit combination, gonadotropin, protein folding, hCG
Reproduction and thyroid function are controlled by the glycoprotein hormones: heterodimeric proteins composed of an α-subunit common to all and a hormone-specific β-subunit (Pierce and Parsons 1981). The free subunits are essentially inactive but can be recombined to form a heterodimer having the activity of the hormone from which the β-subunit is derived (Pierce and Parsons 1981). The tertiary structure of deglycosylated hCG (Lapthorn et al. 1994; Wu et al. 1994) showed that both subunits have a similar topology. Three of the disulfides in each subunit form a cystine knot core that results in an elongated protein containing two paired loops at one end (loops 1 and 3) and a single loop at the other. In the heterodimer, the subunits are oriented in an antiparallel fashion with the single loop of each subunit located adjacent to the paired loops of the other subunit. The β-subunit contains 20 additional amino acids that are wrapped around α-subunit loop 2 like a seatbelt and latched to β-subunit loop 1 by a disulfide (Lapthorn et al. 1994).
The seatbelt makes an obvious contribution to the stability of the heterodimer, yet its location would appear to impede subunit combination. If the seatbelt remained latched, heterodimer formation would occur only if α2 and its associated oligosaccharide are threaded through the small opening between the seatbelt and β1. This requirement would be bypassed if the seatbelt were to open before subunit combination. Protein disulfide isomerase (Huth et al. 1993) and small amounts of reducing agents (Slaughter et al. 1995) accelerate subunit combination in vitro, possibly by breaking the seatbelt latch disulfide. While it might be expected that the seatbelt latch is stable during subunit combination in the absence of these agents, the observation that glycoprotein hormones have an intrinsic thioredoxin activity (Boniface and Reichert 1990; Chew et al. 1995) raised the possibility that docking of their subunits leads to a transient break in the latch disulfide needed for the seatbelt to wrap itself around α2. Following this process, the terminal cysteine of the seatbelt would be in a position to reestablish the seatbelt disulfide, thereby stabilizing the heterodimer. Ruddon et al. (1996) have suggested that the hCG subunits combine before the seatbelt is latched during hCG synthesis in choriocarcinoma and transfected CHO cells. Thus, there is precedent for the possibility that the unlatched seatbelt is wrapped around α2 during the combination process.
Glycoprotein hormone synthesis remains poorly understood in spite of its potential importance as a source of hCG metabolites observed during ectopic pregnancy, Down's syndrome, and even some cancers (Hussa 1987; Cardwell et al. 1997; Cole et al. 1997; Lee et al. 1997). Therefore, we have begun to study this process in greater detail. As described here, we found that Cys 110 was not labeled with alkylating agents during subunit combination, a process that occurs efficiently in oxidizing conditions in vitro. This observation suggested that the unlatching of the seatbelt is not required for subunit combination. Further, none of the cysteines in the β-subunit were alkylated during subunit combination in the presence of a vast molar excess of several alkylating agents. This suggests that breaking of disulfides is not required for subunit combination; this led us to devise a model of heterodimer formation in which movements of the seatbelt and α2 allow this loop and its associated oligosaccharide to pass through the small hole between the seatbelt and the remainder of the β-subunit.
Results and discussion
The seatbelt latch disulfide between Cys 26 and Cys 110 has been shown to be essential for the formation and/or stability of the hCG heterodimer (Suganuma et al. 1989). If the Cys 26–Cys 110 disulfide opened during subunit combination, we anticipated that alkylation of either cysteine would prevent the seatbelt from latching, thereby blocking stable subunit combination. To test this, we monitored subunit combination in the presence of 100 mM VP, 50 mM NEM, 10 mM MB, and 50 and 600 mM IA. We also tested combination in the presence of 1 and 10 mM oxidized glutathione or cystamine. In the absence of alkylating agents, subunit combination was nearly complete within 2 h as analyzed by Western blotting (Fig. 1 ▶) The kinetics of subunit combination were similar to those observed by Ingham et al. (Aloj et al. 1973; Ingham et al. 1973) and by Strickland and Puett (1982). With the exception of IA, which inhibited subunit combination somewhat (Table 1), none of the alkylating agents disturbed heterodimer formation, even when present at 10–1000-fold molar excess (Table 1; Fig. 2 ▶) Combination was not influenced by glutathione or cystamine, agents that would be expected to trap disulfide bonds that had opened during the combination process (Table 2).
Fig. 1.

(Left) Western blot obtained with an anti-α-subunit antibody A113 showing the formation of heterodimer and loss of free α-subunit during combination. (Right) Western blot obtained with anti-β-subunit antibody B112 showing formation of heterodimer and loss of free β-subunit during combination. Note that subunit combination appeared to be nearly quantitative under these conditions.
Table 1.
Effects of alkylating reagents on subunit combination
| Reagent (concentration) | Combination |
| No Alkylating Agent | 100% |
| 4-vinylpyridine (0.1 M) | 102.4% |
| 3-(N-maleimidylpropionyl) biocytin (0.05 M) | 105.1% |
| N-ethylmaleimide (0.05 M) | 101.2% |
| iodoacetate (0.05 M) | 69.2% |
| iodoacetate (0.6 M) | 31.0% |
Subunit combination was measured in the presence of the indicated alkylating agents. The concentrations of alkylating agents were far in excess of that of the subunits (0.1 mM). The only alkylating agent that reduced subunit combination was IA. As discussed, this effect was most likely due to the reaction of IA with noncysteine residues.
Fig. 2.

Time course of subunit combination in vitro in the presence and absence of VP. The amount of heterodimer formed was monitored using a sandwich immunoassay with anti-α-subunit antibody A113 for capture and radioiodinated anti-β-subunit antibody B112 for detection.
Table 2.
Effects of oxidizing agents on subunit combination
| Agent (concentration) | Combination (1 h) |
| None | 100% |
| Oxidized glutathione (1 mM) | 114.3% |
| Oxidized glutathione (10 mM) | 115.9% |
| Cystamine (1 mM) | 124.6% |
| Cystamine (10 mM) | 112.5% |
Subunit combination was measured in the presence of the indicated oxidizing agents. The concentration of the subunits was 0.1 mM.
Conceivably, subunit combination requires disruption of disulfides other than the seatbelt latch.
Disruption of the disulfide between the tenth (Cys 93) and eleventh (Cys 100) cysteines would increase the space between the seatbelt and the β subunit core. This disulfide, which stabilizes a small loop within the seatbelt, is also known to be reduced readily (Pierce and Parsons, 1981). We monitored the alkylation of specific cysteines using MALDI-TOF.
Following subunit combination in the presence of IA or VP, we reduced the heterodimer with 100 mM DTT: a treatment sufficient to cause complete reduction of both subunits (Pollak et al. 1990). The free thiols produced were reacted with an alkylating agent that had a different molecular weight than that used during subunit combination, enabling us to distinguish fragments that had been alkylated before and after reduction. The protein was then digested with N-glycanase to remove the oligosaccharides and with trypsin to produce fragments suitable for MALDI-TOF analysis. We were unable to detect all the expected tryptic fragments of hCG using MALDI-TOF, a phenomenon that is not unique to hCG and may have been related to differences in the abilities of the tryptic peptides to be desorbed from the matrix (Landry et al. 2000). In general, hCG peptides containing cysteines that had been alkylated by VP were much more readily detected than those alkylated by IA. As summarized in Figure 4 ▶, we were able to characterize at least one cysteine from all but two of the 11 disulfides in hCG following reduction, labeling with either IA or VP, and trypsin digestion. The peptide that contained Cys 100 was observed by MALDI-TOF when this cysteine was alkylated by VP but not by IA. Thus, we were able to monitor alkylation of this cysteine only with VP. Neither Cys 93 nor Cys 100 became alkylated during combination in the presence of 100mM VP (Fig. 3 ▶, peak 8). Cys 93 was not alkylated during combination in the presence of 100 mM IA (data not shown). Presumably, Cys 100 was also not alkylated under this condition, but it was not possible to determine this because of the fact that we were unable to observe this peptide when it was labeled with IA. None of the other cysteines detected by mass spectrometry was found to be alkylated by either IA or VP in any trypsin fragment of either subunit (Fig. 3 ▶). These observations suggested that within the limits of the technique, no disulfide bonds had been broken during subunit combination in oxidizing conditions.
Fig. 4.
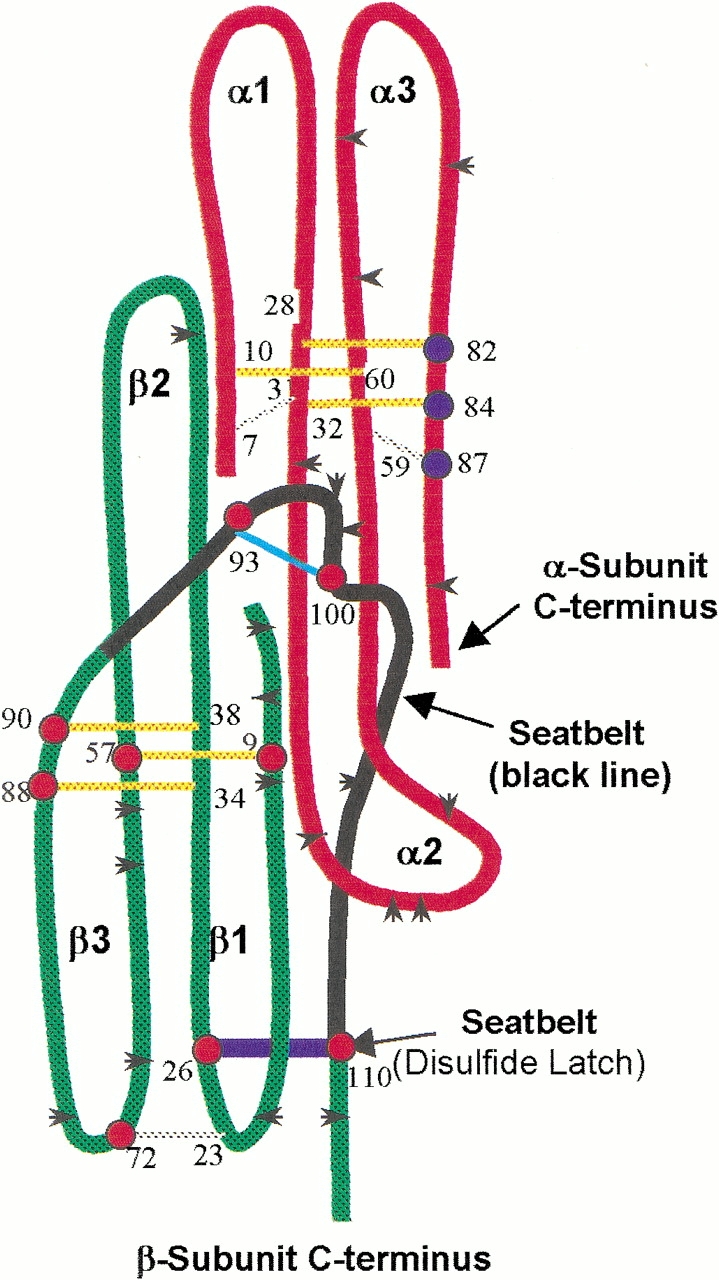
Cysteine residues studied by mass spectrometry. At least one cysteine member of all disulfides in both subunits except α7–α31 and α10–α60 were identified in tryptic peptides by mass spectrometry (α-subunit cysteines, blue dots; β-subunit cysteines, red dots). As summarized from five separate experiments, these cysteines were alkylated only after reduction, not during subunit combination. Thus, disruption of these disulfides was not required for subunit combination. Color scheme: black arrowheads, trypsin cleavage sites; red, α-subunit core; green, β-subunit core; black, β-subunit seatbelt; yellow, cysteine knot disulfides; cyan, seatbelt loop disulfide (Cys 93–Cys 100); blue, seatbelt latch disulfide (Cys 26–Cys 110); gray, remaining disulfides.
Fig. 3.

Cysteine residues identified by MS following reduction and alkylation of hCG. hCG subunit combination was carried out in the presence of VP, and the proteins then were reduced and alkylated by IA (VP > IA). Modification by VP would identify cysteines in disulfides that had broken during subunit combination. After subunit combination had been completed, the proteins were reduced and cysteine residues in disulfides that had not broken during subunit combination were alkylated with IA. As seen here, there was no VP incorporation into cysteine residues during subunit combination. This study was repeated in this and in a reciprocal fashion (not shown) during which IA was used to detect breaking of cysteines during subunit combination and VP was used to label all cysteines after the proteins had been completely reduced (IA > VP). In neither case did we observe alkylation of cysteines during subunit combination. The large spectrum was obtained by MALDI-TOF; the inset shows a tandem mass spectrum obtained for peak number 4. The table illustrates the amino acid sequences of all peptides labeled on the MALDI-TOF spectrum that were identified by tandem mass spectrometry.
We had expected that the oligosaccharide on α-subunit loop 2 would prevent subunit combination because of its bulk (Weisshaar et al. 1991) and that dimer formation could not occur without disruption of the seatbelt latch disulfide. The observations just described indicated that this notion was incorrect. We were concerned that the methods used might not have been sufficient to detect transient disruption of the seatbelt latch or other disulfide, and we examined this possibility. First, we tested the abilities of massive concentrations of IA to inhibit subunit combination. At 600 mM, significant amounts of IA were incorporated into the tyrosine residue of Ala 36–Tyr-Pro-Thr-Pro-Leu–Arg 42, a peptide derived from α-subunit loop 2, and the methionine residue of Val 68–Thr-Val-Met-Gly-Gly-Phe–Lys 75, a peptide derived from α-subunit loop 3, even though each lacks cysteine (Fig. 5 ▶). IA was not incorporated into Cys 93 or Cys 110, β-subunit residues that should have been alkylated more readily than either of these α-subunit peptides, had either the Cys 93–Cys 100 and Cys 26–Cys 110 disulfides been reduced. Residues in α-subunit loop 2 near its interface with the β-subunit have been shown to reduce subunit combination (Bielinska and Boime 1992a, b, Grossmann et al. 1996). Modification of Tyr 37 would explain the ability of massive amounts of IA to reduce subunit combination without appearing to alter the seatbelt latch or other disulfide (Table 1).
Fig. 5.

Incorporation of IA into residues other than cysteine. hCG α-subunit was incubated with 50mM IA (pH 7.8, 37°C, 4 h), purified by adsorption to C18 resin in zip-tips, and digested with trypsin. Peptides Ala-Tyr-Pro-Thr-Pro-Leu-Arg from α2 (peak 1) and Val-Thr-Val-Met-Gly-Gly-Phe-Lys from α3 (peak 2) were found to be modified by IA (peaks 3 and 4, respectively). The sequence of peak 3 was found to be Ala-Tyr(IA)-Pro-Thr-Pro-Leu-Arg. Peak 4 was not characterized further.
Cys 26 is part of a large tryptic fragment that contains several cysteines (i.e., Cys 23, Cys 26, Cys 34, Cys 38) that was not detected following treatment with IA or VP using the alkylation/MALDI-TOF procedure just described. We were able to study the reactivity of Cys 26 using biotinylated alkylating agents and a monoclonal antibody (i.e., B111) that recognizes a β-subunit epitope that includes the seatbelt latch disulfide (Cosowsky et al. 1995).
Biotinylation of Cys 26 or Cys 110 by these alkylating agents would lead to a molecule that binds streptavidin and monoclonal antibodies to other parts of the β-subunit (e.g., B110), but it also would prevent binding of B111. Alkylation of cysteines or other residues is readily detected in sandwich immunoassays using streptavidin for capture and radioiodinated antibodies for detection or vice versa. Because of the high affinities of streptavidin for biotin and the high specific activities to which the antibodies can be radiolabeled, this assay is much more sensitive than the MALDI-TOF method.
MB failed to inhibit subunit combination (Fig. 6 ▶, upper panels), even though it was incorporated into the heterodimer (Fig. 6 ▶, lower panels) and both free subunits (Fig. 7 ▶). Under these conditions, however, MB was not incorporated into β-subunit Cys 26 or Cys 110 as assessed with a Western blot streptavidin-sandwich assay. In this assay, the proteins were captured by streptavidin that had been adsorbed to the nitrocellulose and detected with anti-β-subunit antibodies B110 and B111. The latter is sensitive to the presence of the seatbelt latch disulfide. As seen (Fig. 6 ▶, lower panels), the biotinylated β-subunit and heterodimer were detected nearly equally by B110 and B111. Thus, only trace amounts of biotin, if any, were associated with either Cys 26 or Cys 110. Furthermore, it was possible to drive all of the β-subunit into the heterodimer by increasing the amount of α-subunit in the reaction mixture (Fig. 6 ▶, all panels). Alkylation of either Cys 26 or Cys 110 in even a fraction of the β-subunit would have led to detectable amounts of free β-subunit in the streptavidin-B110 assay. We have not attempted to identify residues in either subunit that reacted with MB but have observed that trypsin digestion produced a peptide fragment having a molecular weight of 1340.9 (not shown). This is consistent with biotinylation of Tyr 37 in the sequence Ala 36–Tyr-Pro-Thr-Pro-Leu–Arg 42, a peptide derived from α-subunit loop 2. This peptide had also been alkylated by IA (Fig. 5 ▶).
Fig. 6.

Western blots illustrating the results of subunit combination in the presence of 10 mM MB. hCG subunit combination was studied overnight at 37°C at different molar ratios of α- and β-subunit in the presence or the absence of 10 mM MB. The heterodimer was separated from the free subunits by electrophoresis and the proteins transferred to nitrocellulose (A) or streptavidin-coated nitrocellulose (B). The former captures all proteins and the latter captures only those proteins that are biotinylated. Free β subunit was used as control. B111 binding, which requires formation of the seatbelt disulfide, was comparable to B110 binding in all conditions. Some MB was incorporated into the free β-subunit, showing that the presence of the α-subunit was not required for this effect. The ability of the free β-subunit to bind B111 comparable to B110 showed that the seatbelt latch was not broken under these conditions.
Fig. 7.

Incorporation of biotin into both subunits during combination in the presence of 10 mM MB. Following combination of equimolar amounts of α- and β-subunits in the presence of 10 mM MB, half the reaction mixture was treated with 10 M urea at pH 4.5 to separate the subunits (lane 2); the heterodimer and free subunits were separated by electrophoresis and blotted onto nitrocellulose. Biotinylated proteins were detected using streptavidin linked horseradish peroxidase. As can be seen, both subunits were alkylated by MB. In addition, the heterodimer was dissociated completely by urea treatment, an indication that no intersubunit disulfide had formed during subunit combination.
The rate of subunit combination has been shown to be increased by several-fold in mild reducing conditions (Slaughter et al. 1995). This might be explained by opening of the seatbelt latch disulfide, one of the more easily reduced disulfides in the glycoprotein hormones (Pierce and Parsons 1981), a process expected to be highly sensitive to the presence of alkylating agents. In confirmation of earlier studies, we found that subunit combination occurred sixfold more rapidly in the presence of 1 mM β-mercaptoethanol than in its absence (Fig. 8 ▶). Higher concentrations of β-mercaptoethanol were less effective in promoting subunit combination (not shown), and when present at 10 mM, β-mercaptoethanol prevented subunit combination, most likely by denaturing the subunits. Even at 1 mM, β-mercaptoethanol caused the reduction of several disulfides, not just the seatbelt latch. We detected alkylation of Cys 72, Cys 93, Cys 100, and Cys 110 by 10 mM VP when subunit combination had been initiated in the presence of 1 mM β-mercaptoethanol (Fig. 9 ▶). Thus, in addition to destabilizing the seatbelt latch (Cys 26–Cys 110), mild reduction altered the disulfide between β-subunit loops 1 and 3 (Cys 23–Cys 72) and the disulfide within the small seatbelt loop (Cys 93–Cys 100). Reduction of these disulfides in 1 mM β-mercaptoethanol was only partial, as shown by the finding that complete reduction of the β-subunit with 100 mM DTT and alkylation with IA modified Cys 72, Cys 93, and Cys 110, as well as cysteines that had not been modified by VP (Fig. 10 ▶). Unlike subunit combination in oxidizing conditions, which was not blocked by 100–1000-fold molar excess of alkylating agent, subunit combination during mildly reducing conditions was inhibited immediately and quantitatively by a 10-fold molar excess of alkylating agent (Fig. 10 ▶). These observations suggested that we would have observed alkylation of the cysteines in the seatbelt latch had this disulfide been broken during subunit combination under oxidizing conditions, particularly as VP and IA modified peptides were readily resolved during MALDI-TOF (Fig. 9 ▶).
Fig. 8.

Subunit combination in the presence of β-mercaptoethanol. hCG α- and β-subunits were combined in the absence (squares) or presence of 0.1 mM (triangles) and 1 mM (diamonds) β-mercaptoethanol. The reaction rate in the presence of 1 mM β-mercaptoethanol was six times that in its absence. When present at 3 mM, β-mercaptoethanol enhanced subunit combination, but not nearly as well as 1 mM (not shown). At 10 mM, β-mercaptoethanol prevented subunit combination (not shown).
Fig. 9.

Identification of cysteines alkylated by VP during subunit combination in the presence of β-mercaptoethanol. hCG subunits were combined in the presence of 1 mM BME. VP (10 mM) was added within 5–10 sec after combination reaction had been initiated. Four hours later, when subunit combination was assumed to be complete, excess VP was removed by electrophoresis on a 15% SDS-polyacrylamide gel and proteins were electroeluted from a strip of gel containing the heterodimer and free subunits. Following denaturation and reduction of these proteins, cysteines that had not been alkylated by VP during subunit combination were alkylated by IA. Peptides produced by N-glycanase and trypsin digestion were subjected to MALDI-TOF to distinguish those that contained cysteines labeled by VP and IA. Shown here is a typical mass spectrum with the inset illustrating an enlarged region near peak 5. Highlighted amino acids in the table were labeled by VP or IA as noted. Note that cysteines from disulfides βC23–C72, βC93–C100 and βC26–C100 were labeled by both VP and IA, indicating that a portion of these disulfides had been reduced during subunit combination in the presence of 1 mM β-mercaptoethanol. The sequences of all peptides listed in the table were confirmed by LC/MS/MS.
Fig. 10.

Subunit combination in the presence of 1 mM BME can be blocked by 10 mM IA. Concentrations of IA 50 mM or more were required to inhibit subunit combination in the absence of β-mercaptoethanol (Table 1), an effect that may have been caused by modification of residues other than cysteine (Fig. 5 ▶). Low concentrations of IA (10 mM) completely terminated subunit combination observed in the presence of 1 mM β-mercaptoethanol. Inhibition of combination was observed immediately after addition of IA, however, regardless of whether it was added 5–10 sec (upright triangles, uneven dashes) or 5 min (inverted triangles, even dashes) after subunit combination had been initiated. IA had no influence on heterodimer that had already been formed (inverted triangles, even dashes). In the absence of β-mercaptoethanol, 10 mM IA did not inhibit subunit combination (squares, solid line).
MALDI-TOF is a powerful analytical technique that can be used to identify peptide fragments, but its use as a quantitative tool is limited. In an effort to determine the relative amounts of each cysteine that had been modified during subunit combination in the presence of mild reducing conditions, we trapped cysteines that had been reduced using a fluorescent alkylating agent (IAEDANS). Following trypsin digestion of hCG, it was possible to quantify the relative amounts of each labeled tryptic fragment by HPLC using a fluorescence detector. As seen (Fig. 11 ▶), the AEDANS fluorophore was present in two peptides in nearly equal amounts. MALDI-TOF analyses showed that fluorescent peptides Gly 75–Val-Asn-Pro-Val-Val-Ser-Tyr-Ala-Val-Ala-Leu-Ser-Cys-Gln-Cys-Ala-Leu-Cys–Arg 94 and Ser 96–Thr-Thr-Asp-Cys-Gly-Gly-Pro–Lys 104 each contained one molecule of AEDANS. Though we did not determine the locations of the AEDANS residues in each peptide by tandem mass spectrometry, it is highly probable that these were located on Cys 93, based on the ability of this cysteine in this peptide to be preferentially alkylated following mild reduction (Fig. 9 ▶) and on Cys 100. Only trace amounts of AEDANS were found on any other peptide fragment of hCG, including that which contained Cys 110. This showed that even in the presence of mild reducing conditions, the Cys 26–Cys 110 bond does not appear to be disrupted in conditions that enhance subunit combination.
Fig. 11.

Relative amounts of fluorescent peptides observed following alkylation with IAEDANS. Incorporation of IAEDANS into hCG and its α- and β-subunits was monitored during subunit combination in the presence and absence of β-mercaptoethanol. Only trace amounts of IAEDANS were incorporated into these proteins in the absence of β-mercaptoethanol (not shown). In the presence of 1 and 3 mM β-mercaptoethanol, nearly all the incorporated AEDANS fluorophore was found equally in two peptides, namely β–Gly 75–Val-Asn-Pro-Val-Val-Ser-Tyr-Ala-Val-Ala-Leu-Ser-Cys-Gln-Cys-Ala-Leu-Cys–Arg 94 (peak 1) and β–Ser 96–Thr-Thr-Asp-Cys-Gly-Gly-Pro–Lys 104 (peak 2). The data shown here were obtained in the presence of 3 mM β-mercaptoethanol. MALDI-TOF analyses showed that only one molecule of AEDANS had been incorporated into each peptide. This is consistent with the idea that mild reduction disrupted the disulfide between the small seatbelt loop stabilized by β-subunit cysteines Cys 93 and Cys 100, making each available for alkylation by IAEDANS. Only trace amounts of AEDANS were incorporated into Cys 110 as seen by the relative absence of fluorescence in the peptide that contained this cysteine (Asp 105–His-Pro-Leu-Thr-Cys-Asp-Asp-Pro–Arg 114; peak 3). These results indicated that the acceleration of subunit combination at low concentrations of reducing agents was caused by the opening of the small seatbelt loop, a phenomenon that would increase the size of the hole in the β-subunit.
These observations indicate that α-subunit loop 2 and its associated oligosaccharide can pass through the small hole created by latching of the seatbelt to the core of the β-subunit during subunit combination. How can the subunits combine while the seatbelt latch remains intact? We propose that the position of the seatbelt in the free β-subunit differs considerably from that in the heterodimer. Experimental support for this idea comes from the observation that many hCG-specific antibodies (i.e., B107, B109) recognize an epitope composed of β2 and the N-terminal end of the seatbelt (Moyle et al. 1990). These antibodies have very low ability to bind the free β-subunit, indicating that this epitope has a different conformation in hCG and in the free subunit. When present in a position such as that in Figure 12A ▶, the seatbelt would not prohibit docking of the α- and β-subunits. In fact, in this position, the seatbelt may contribute to subunit interaction. Following the reversible interactions between the two subunits (Fig. 12B ▶), movements of the seatbelt relative to α2 and its associated oligosaccharide (Fig. 12C ▶) continue until these portions of each subunit occupy the more stable conformations seen in the heterodimer (Fig. 12D ▶). Loop 2 of the α-subunit is known to be highly mobile (De Beer et al. 1996), a factor that may favor this transition but is not illustrated in Figure 12 ▶.
Fig. 12.

Model for subunit combination under conditions in which the seatbelt remains latched. We propose that the seatbelt can occupy a different conformation in the free subunit and in the heterodimer as illustrated by the black ribbon in the extreme left and right panels. The position of the seatbelt in the free subunit does not interfere with the initial interaction between the two subunits as seen in the second panel. The seatbelt passes over the second α-subunit loop as shown in the third panel to reach the position it occupies in the heterodimer, a process likely to be facilitated by the mobility of α2. The reverse of this process may lead to subunit dissociation. Color scheme: α-subunit, dark thin line; β-subunit, gray thin line; seatbelt, black ribbon. Note that residues from the core portions of the α- and β-subunits are taken from the crystal structure. The locations of the seatbelt in panels a–c are entirely speculative, while that in the last panel is taken from the crystal structure. The structures in panels a–c were subjected to energy minimization to make certain that there were no bad contacts, unusual bond lengths, or disallowed bond angles before the ribbon diagrams were generated.
The observation that small quantities of reducing agents appear to disrupt the Cys 93–Cys 100 disulfide suggests that mild reduction (Slaughter et al. 1995) or protein disulfide isomerase treatment (Huth et al. 1993) can facilitate subunit combination by increasing the opening between the seatbelt and the core of the β-subunit. It remains to be determined if this occurs in living cells, however. These studies suggest that the subunits can combine in vitro while the seatbelt is latched and raise the possibility that this can also occur during synthesis in the endoplasmic reticulum.
Materials and methods
Reagents
Pure recombinant hCG (Ares Serono International) was separated into its constituent subunits by reversed phase HPLC on a Vydac C4 column (22.5 × 250 mm) in 0.1% aqueous TFA with a 15%–40% linear gradient of 0.079% TFA in acetonitrile. The isolated subunits were equilibrated in 10 mM sodium phosphate buffer (pH 7.2) by ultrafiltration on a 5000YM Amicon membrane. The concentration of each subunit was determined in triplicate by automated analysis on an ABI420A amino acid analyzer. Antibodies A113 and B111 were given to us by Glenn Armstrong and Robert Wolfert, Hybritech. B110 was prepared in this laboratory (Moyle et al. 1987). Antibody A113 recognizes an epitope found primarily on the surface of α-subunit loop 1 near its interface with the β-subunit.
Antibody B110 binds an epitope on the convex surface of β-subunit loops 1 and 3 that does not include the seatbelt. Antibody B111 binds an epitope that includes the seatbelt latch disulfide. N-ethylmaleimide (NEM), iodoacetate (IA), iodoacetamide, 4-vinylpyridine (VP), and streptavidin (SA) were purchased from Sigma. 3–(N-maleimidylpropionyl)biocytin (MB), a biotin containing derivative of NEM, and 5–((((iodoacetyl)amino)ethyl)amino)napthalene-1-sulfonic acid (IAEDANS) were purchased from Molecular Probes. Antibodies used for detection of free subunits and heterodimers in Western blotting were radioiodinated using an Iodo-Gen-based technique (Cruz et al. 1987).
Subunit combination and analysis of heterodimer
Equimolar concentrations of subunits (0.08–0.1mM) in 25 mM phosphate, 5 mM KCl (pH 7.5) were incubated at 37°C in the presence or absence of high concentrations of alkylating agents as noted in the figure and table legends. As noted also, lower concentrations of alkylating agents were used to study subunit combination that had been initiated in the presence of 1 mM β-mercaptoethanol. Heterodimer formation was measured using sandwich immunoassays (Moyle et al. 1982) and Western blot analysis (Moyle et al. 1990). Free and combined subunits were separated by electrophoresis on 12% polyacrylamide gels containing 0.1% sodium dodecyl sulfate. Proteins electroeluted from the gel onto nitrocellulose sheets were detected using radiolabeled antibodies to either the α-subunit (A113) or β-subunit (B110, B111). Biotinylation of the subunits and the heterodimer by MB was monitored by electrophoresis and Western blotting. The presence of biotin in these proteins was determined by chemiluminescence using the streptavidin-linked horseradish peroxidase kit obtained from Boehringer Mannheim. Incorporation of MB was also monitored by a Western blot–sandwich procedure in which the biotinylated subunits and dimer were separated by electrophoresis and then transferred to nitrocellulose sheets that had been coated with streptavidin (20 μg/mL, 1 h, 37°C) and blocked with bovine serum albumin (1 mg/mL, 1.5 h, room temperature). Following transfer, only those proteins that contained biotin were retained on the membranes. Detection of biotin labeled heterodimer and β subunit was performed with B110 or B111 radiolabeled to a specific activity of 50 μCi/μg.
Analysis of disulfide opening during subunit combination
We combined the subunits for 2–4 h or overnight at 37°C in the presence of 100 mM IA or 100 mM VP to trap cysteines created by the breaking of disulfide bonds during heterodimer formation. Unreacted alkylating agents were removed from the reaction mixture by reversed-phase HPLC on C18 resin (Phenomenex) or by electrophoresis. Following the latter procedure, the heterodimers and subunits were recovered from gel slices by electroeluting them at 40 V overnight. The samples were denatured and all remaining disulfide bonds were disrupted by incubation in an argon-purged solution containing 10 M urea, 2 mM EDTA, 20 mM Tris (pH 7.5), and 100 mM DTT for 1–2 h at 37°C. Cysteines that had not been alkylated during subunit combination in the presence of IA were alkylated in the presence of 200 mM VP (IA→VP). Conversely, subunits that had been exposed to VP during combination were alkylated with 200 mM IA (VP→IA). These alkylations were performed at room temperature for 2–3 h or overnight at 4°C. The denatured and alkylated proteins were again purified by reversed-phase HPLC or by "zip-tips" containing C18 resin (Millipore) to remove salts, urea, and unreacted VP or IA. After being lyophilized, the dried proteins were dissolved in 50 mM Tris-HCl buffer (pH 7.5) containing 1 mM CaCl2 and digested with TPCK-treated trypsin (Promega) at 37°C overnight.
In some cases, the oligosaccharides were removed before trypsin digestion by treating the samples with N-glycanase (New England Biolabs), as recommended by the supplier except for the omission of NP40. The tryptic digests were analyzed by MALDI/TOF mass spectrometry using the Voyager DE/STR and DE/PRO instruments (PE Biosystems) operated in reflector mode using delayed extraction. Each spectrum was externally calibrated using a mixture of standard peptides. The identities of these peptides were confirmed following electrospray ionization using an LCQ ion trap mass spectrometer (Finnigan–MAT) operated in automated LC/MS/MS mode.
Analysis of disulfide opening using IAEDANS: hCG α- and β-subunits (0.05 mM) were combined in the presence of IAEDANS (5 mM) with 0, 1, and 3 mM β-mercaptoethanol at 37°C for 2 h. The combined and free subunits were separated from unreacted IAEDANS by electrophoresis through 0.1% dodecylsulfate-12% polyacrylamide gels, electroeluted from the gels in running buffer diluted 1 : 10, and mixed.
Following lyophyllization, they were reduced by 100 mM DTT and alkylated with 250 mM iodoacetamide. The alkylated subunits were purified by reversed phase HPLC on a C18 resin and digested with trypsin. The fragments were separated by reversed phase HPLC on C18 using a gradient of 0%–95% acetonitrile in 10 mM ammonium bicarbonate (pH 8). Fluorescence was monitored at 490 nm following 337 nm excitation.
Acknowledgments
These studies were supported by NIH grants HD14907, HD38547, and DK50600. We thank Robert Wolfert and Glenn Armstrong for antibodies and Roberta Napoleoni for the amino acid analysis determinations.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
hCG, human chorionic gonadotropin
NEM, N-ethylmaleimide
IA, iodoacetate
VP, 4-vinylpyridine
SA, streptavidin
MB, 3–(N-maleimidylpropionyl)biocytin
IAEDANS, 5–((((2-iodoacetyl)amino)ethyl)amino)napthalene-1-sulfonic acid.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.25901.
References
- Aloj, S.M., Edelhock, H., Ingham, K.C., Morgan, F.J., Canfield, R.E., Ross, G.T. 1973. The rates of dissociation and reassociation of the subunits of human chorionic gonadotropin. Arch. Biochem. Biophys. 159 497–504. [DOI] [PubMed] [Google Scholar]
- Bielinska, M. and Boime, I. 1992a. Site-directed mutagenesis defines a domain in the gonadotropin α-subunit required for assembly with the chorionic gonadotropin β-subunit. Mol. Endocrinol. 6 261–271. [DOI] [PubMed] [Google Scholar]
- Bielinska, M. and Boime, I. 1992b. Site-directed mutagenesis defines a domain in the gonadotropin α-subunit required for assembly with the chorionic gonadotropin β-subunit. Mol. Endocrinol. 6 267–271. [DOI] [PubMed] [Google Scholar]
- Boniface, J.J. and Reichert, L.E.J. 1990. Evidence for a novel thioredoxin-like catalytic property of gonadotropic hormones. Science 247 61–64. [DOI] [PubMed] [Google Scholar]
- Cardwell, L., Kowalczyk, C.L., Krivchenia, E.L., Leon, J., and Evans, M.I. 1997. Urinary β-core fragment as a predictor of abnormal pregnancy at 4–6 weeks' gestation. Fetal Diagn. Ther. 12 340–342. [DOI] [PubMed] [Google Scholar]
- Chew, C.C., Magallon, T., Martinat, N., Lecompte, F., Combarnous, Y., and Gosling, J.P. 1995. The relative protein disulphide isomerase (PDI) activities of gonadotrophins, thioredoxin and PDI. Biochem. Soc. Trans. 23 394S. [DOI] [PubMed] [Google Scholar]
- Cole, L.A., Isozaki, T., and Jones, E.E. 1997. Urine β-core fragment, a potential screening test for ectopic pregnancy and spontaneous abortion. Fetal Diagn. Ther. 12 336–339. [DOI] [PubMed] [Google Scholar]
- Cosowsky, L., Rao, S.N.V., Macdonald, G.J., Papkoff, H., Campbell, R.K., and Moyle, W.R. 1995. The groove between the α- and β-subunits of hormones with lutropin (LH) activity appears to contact the LH receptor and its conformation is changed during hormone binding. J. Biol. Chem. 270 20011–20019. [DOI] [PubMed] [Google Scholar]
- Cruz, R.I., Anderson, D.M., Armstrong, E.G., and Moyle, W.R. 1987. Nonreceptor binding of human chorionic gonadotropin (hCG): Detection of hCG or a related molecule bound to endometrial tissue during pregnancy using labeled monoclonal antibodies that bind to exposed epitopes on the hormone. J. Clin. Endocrinol. Metab. 64 433–440. [DOI] [PubMed] [Google Scholar]
- De Beer, T., Van Zuylen, C.W., Leeflang, B.R., Hard, K., Boelens, R., Kaptein, R., Kamerling, J.P., and Vliegenthart, J.F. 1996. NMR studies of the free α subunit of human chorionic gonadotropin: Structural influences of N-glycosylation and the β subunit on the conformation of the α subunit. Eur. J. Biochem. 241 229–242. [DOI] [PubMed] [Google Scholar]
- Grossmann, M., Szkudlinski, M.W., Dias, J.A., Xia, H., Wong, R.P., and Weintraub, B.D. 1996. Site-directed mutagenesis of amino acids 33–44 of the common α-subunit reveals different structural requirements for heterodimer expression among the glycoprotein hormones and suggests that cyclic adenosine 3`,5`-monophosphate production and growth promotion are potentially dissociable functions of human thyrotropin. Mol. Endocrinol. 10 769–779. [DOI] [PubMed] [Google Scholar]
- Hussa, R.O. 1987. The clinical marker hCG. Praeger, New York.
- Huth, J.R., Perini, F., Lockridge, O., Bedows, E., and Ruddon, R.W. 1993. Protein folding and assembly in vitro parallel intracellular folding and assembly: Catalysis of folding and assembly of the human chorionic gonadotropin α β dimer by protein disulfide isomerase. J. Biol. Chem. 268 16472–16482. [PubMed] [Google Scholar]
- Ingham, K.C., Aloj, S.M., and Edelhoch, H. 1973. The rates of dissociation and recombination of the subunits of luteinizing hormone. Arch. Biochem. Biophys. 159 596–605. [DOI] [PubMed] [Google Scholar]
- Landry, F., Lombardo, C.R., and Smith J.W. 2000. A method for application of samples to matrix-assisted laser desorption ionization time-of-flight targets that enhances peptide detection. Anal. Biochem. 279 1–8. [DOI] [PubMed] [Google Scholar]
- Lapthorn, A.J., Harris, D.C., Littlejohn, A., Lustbader, J.W., Canfield, R.E., Machin, K.J., Morgan, F.J., and Isaacs, N.W. 1994. Crystal structure of human chorionic gonadotropin. Nature 369 455–461. [DOI] [PubMed] [Google Scholar]
- Lee, I.S., Chung, D.Y., Cole, L.A., Copel, J.A., Isozaki, T., and Hsu, C.D. 1997. Elevated serum nicked and urinary β-core fragment hCG in preeclamptic pregnancies. Obstet. Gynecol. 90 889–892. [DOI] [PubMed] [Google Scholar]
- Moyle, W.R., Ehrlich, P.H., and Canfield, R.E. 1982. Use of monoclonal antibodies to hCG subunits to examine the orientation of hCG in the hormone-receptor complex. Proc. Natl. Acad. Sci. 79 2245–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle, W.R., Pressey, A., Dean Emig, D., Anderson, D.M., Demeter, M., Lustbader, J., and Ehrlich, P. 1987. Detection of conformational changes in human chorionic gonadotropin upon binding to rat gonadal receptors. J. Biol. Chem. 262 16920–16926. [PubMed] [Google Scholar]
- Moyle, W.R., Matzuk, M.M., Campbell, R.K., Cogliani, E., Dean Emig, D.M., Krichevsky, A., Barnett, R.W., and Boime, I. 1990. Localization of residues that confer antibody binding specificity using human chorionic gonadotropin/luteinizing hormone β subunit chimeras and mutants. J. Biol. Chem. 265 8511–8518. [PubMed] [Google Scholar]
- Pierce, J.G. and Parsons, T.F. 1981. Glycoprotein hormones: Structure and function. Annu. Rev. Biochem. 50 465–495. [DOI] [PubMed] [Google Scholar]
- Pollak, S., Halpine, S., Chait, B.T., and Birken, S. 1990. High resolution high performance liquid chromatography fingerprinting of purified human chorionic gonadotropin demonstrates that oxidation is a cause of hormone heterogeneity. Endocrinol. 126 199–208. [DOI] [PubMed] [Google Scholar]
- Ruddon, R.W., Sherman, S.A., and Bedows, E. 1996. Protein folding in the endoplasmic reticulum: Lessons from the human chorionic gonadotropin β-subunit. Prot. Sci. 8 1443–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter, S., Wang, Y.H., Myers, R.V., and Moyle, W.R. 1995. The lutropin β-subunit N-terminus facilitates subunit combination by offsetting the inhibitory effects of residues needed for LH activity. Mol. Cell. Endocrinol. 112 21–25. [DOI] [PubMed] [Google Scholar]
- Strickland, T.W. and Puett, D. 1982. The kinetic and equilibrium parameters of subunit association and gonadotropin dissociation. J. Biol. Chem. 257 2954–2960. [PubMed] [Google Scholar]
- Suganuma, N., Matzuk, M.M., and Boime, I. 1989. Elimination of disulfide bonds affects assembly and secretion of the human chorionic gonadotropin β subunit. J. Biol. Chem. 264 19302–19307. [PubMed] [Google Scholar]
- Weisshaar, G., Hiyama, J., and Renwick, A.G. 1991. Site-specific N-glycosylation of human chorionic gonadotrophin—Structural analysis of glycopeptides by one- and two-dimensional 1H NMR spectroscopy. Glycobiology 1 393–404. [DOI] [PubMed] [Google Scholar]
- Wu, H., Lustbader, J.W., Liu, Y., Canfield, R.E., and Hendrickson, W.A. 1994. Structure of human chorionic gonadotropin at 2.6 Å resolution from MAD analysis of the selenomethionyl protein. Structure 2 545–558. [DOI] [PubMed] [Google Scholar]