

Abstract
The repressor proteins of the LacI/GalR family exhibit significant similarity in their secondary and tertiary structures despite less than 35% identity in their primary sequences. Furthermore, the core domains of these oligomeric repressors, which mediate dimerization, are homologous with the monomeric periplasmic binding proteins, extending the issue of plasticity to quaternary structure. To elucidate the determinants of assembly, a structure-based alignment has been created for three repressors and four periplasmic binding proteins. Contact maps have also been constructed for the three repressor interfaces to distinguish any conserved interactions. These analyses show few strict requirements for assembly of the core N-subdomain interface. The interfaces of repressor core C-subdomains are well conserved at the structural level, and their primary sequences differ significantly from the monomeric periplasmic binding proteins at positions equivalent to LacI 281 and 282. However, previous biochemical and phenotypic analyses indicate that LacI tolerates many mutations at 281. Mutations at LacI 282 were shown to abrogate assembly, but for Y282D this could be compensated by a second-site mutation in the core N-subdomain at K84 to L or A. Using the link between LacI assembly and function, we have further identified 22 second-site mutations that compensate the Y282D dimerization defect in vivo. The sites of these mutations fall into several structural regions, each of which may influence assembly by a different mechanism. Thus, the 360-amino acid scaffold of LacI allows plasticity of its quaternary structure. The periplasmic binding proteins may require only minimal changes to facilitate oligomerization similar to the repressor proteins.
Keywords: Structural plasticity, quaternary structure, lactose repressor protein, purine repressor protein, periplasmic binding protein, trehalose repressor protein
One fascinating aspect of protein folding is that a wide range of primary sequences can generate very similar secondary and tertiary structures. For example, Thornton and her colleagues have demonstrated that many sequences with less than 35% identity have the same folding topology (Swindells et al. 1998; this reference also provides a very helpful review of the various structural classification schemes currently available). Plasticity of folding is well illustrated by the LacI/GalR family of repressor proteins.† The breadth of this family has been recognized (von Wilcken-Bergmann and Müller-Hill 1982; Weickert and Adhya 1992), and a recent search of protein sequence databases found more than 30 repressor proteins from various bacterial strains with similarity to LacI (Swiss Institute of Bioinformatics BLAST search, Altschul et al. 1990). Of these, PurR has the highest sequence identity with LacI, ∼32%. Furthermore, the core domain of LacI has 10%–25% sequence identity with periplasmic sugar binding proteins (Müller-Hill 1983; Sams et al. 1984; Mowbry and Björkman 1999).
Predictions of similar tertiary structures between and within these two protein families were validated by comparing the X-ray crystallographic structures of LacI, PurR, and TreR (Schumacher et al. 1994, 1995; Friedman et al. 1995; Lewis et al. 1996; Hars et al. 1998; Bell and Lewis 2000) and GBP, RBP, ABP, and ALBP (Newcomer et al. 1981; Quiocho and Vyas 1984; Vyas et al. 1988, 1991; Mowbray and Cole 1992; Björkman and Mowbray 1998; Chaudhuri et al. 1999). The repressor core domains and the binding proteins consist of two subdomains connected by three strands that cross back and forth between them (see LacI structure in Fig. 1 ▶) Each subdomain has a central β sheet surrounded by α helices, and the sugar/effector binding site (Fig. 1 ▶, *) is formed by the residues that line a cleft between the two subdomains (Newcomer et al. 1981; Quiocho and Vyas 1984; Vyas et al. 1988, 1991; Mowbray and Cole 1992; Schumacher et al. 1994, 1995; Friedman et al. 1995; Lewis et al. 1996; Björkman and Mowbray 1998; Hars et al. 1998; Chaudhuri et al. 1999; Bell and Lewis 2000). In light of the striking structural similarities of these proteins, the difference in their assembly states is conspicuous: The repressor proteins all form oligomers, with the monomer–monomer interface located on the core domains (Weickert and Adhya 1992; Schumacher et al. 1994, 1995; Friedman et al. 1995; Lewis et al. 1996; Hars et al. 1998; Bell and Lewis 2000), whereas the periplasmic sugar binding proteins are monomeric. This observation raises questions of what determinants are required for LacI/GalR family assembly and whether and how the concept of plasticity in folding can be extended to quaternary structure. A structure-based alignment of three repressors and four periplasmic binding proteins has been created to examine these issues.
Fig. 1.

Structure of LacI. The LacI homotetramer is best described as a dimer of dimers (Friedman et al. 1995; Lewis et al. 1996). Monomers of the right dimer are black and gray to show how the N-terminal DNA-binding domains "cross over" the core domains of their partners. DNA is indicated at the top of the figure with balls and sticks. Each monomer comprises 360 amino acids. The first 60 amino acids are involved in DNA binding (Geisler and Weber 1977; Jovin et al. 1977; Ogata and Gilbert 1978; Khoury et al. 1991); a complete DNA binding site is composed of the two DNA-binding domains of a dimer (dark gray on left dimer). Residues 61–340 make up the core domain (light gray on left dimer; Platt et al. 1973; Files and Weber 1976), which contains the inducer binding site (one per monomer) and the monomer–monomer interface. The two inducer binding sites of the left dimer are indicated (*). The remaining residues (341–360) encompass an LHR sequence that forms the tetramerization domain (bottom of figure; Alberti et al. 1991, 1993; Chakerian et al. 1991; Chen et al. 1992a). This structure was generated from the pdb file 1lbg (Lewis et al. 1996).
Interestingly, wild-type LacI itself shows some quaternary structural plasticity: Two structures have been determined for tetrameric, inducer-bound LacI: that of only the core (Friedman et al. 1995) and that of complete protein (Lewis et al. 1996). The four monomer–monomer interfaces in these structures all differ slightly, primarily in the number of atomic contacts made by various residues. Most contacts between LacI monomers are mediated through one face of the core domain (Fig. 2 ▶) Additional monomer–monomer contacts occur between the DNA binding domain and core N-subdomain and between the core C-subdomain and the LHR (Fig. 1 ▶).
Fig. 2.
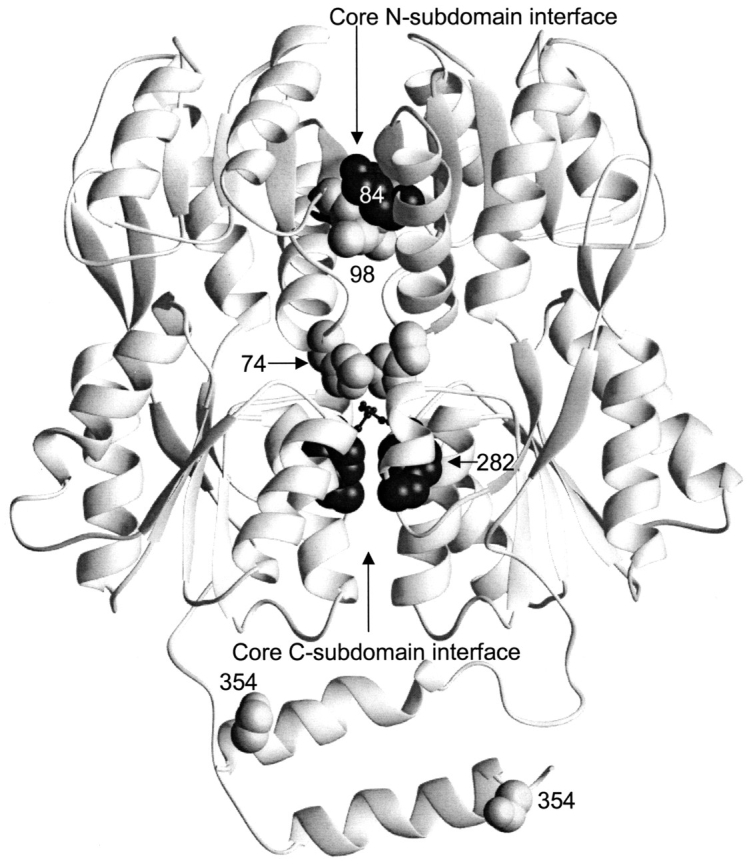
Mutations in the monomer–monomer interface of LacI. This structure depicts one of the two dimers of the IPTG-bound structure of LacI (1lbh; Lewis et al. 1996). The N-terminal DNA binding domain is not resolved for this structure. Mutations affecting interface strength and assembly (at positions 84, 98, 74, 282, and 354) are indicated with spheres. D278, which forms an ion pair with H74, is indicated with the smaller balls and sticks.
Contributions to dimerization from the core C-subdomain have been known to be important for a long time. The significance of the region around 282 was indicated as early as 1976 when the variant designated T41 was determined to be monomeric (Schmitz et al. 1976). The sequence of this mutant was later found to contain the point mutation Y282D (Fig. 2 ▶; Chen and Matthews 1992b). In vitro characterization of purified Y282D demonstrated that the monomeric protein was folded and exhibited wild-type inducer binding properties (Daly and Matthews 1986). Later in vitro experiments showed that substituting A, E, and S at this position also disrupted assembly, whereas L and F did not (Chakerian and Matthews 1991). Changes at LacI 281 are not as dramatic but can also compromise assembly (Fig. 2 ▶; Chakerian and Matthews 1991; Spott et al. 2000). Although neither single mutation abrogates assembly, the double mutation C281S/Y282L does (Chakerian and Matthews 1991). Combinations of mutations at 281 and other sites such as 223, 255, 282, and 283 also compromise assembly as measured by phenotypic behavior (Spott et al. 2000). A number of other mutations near the core C-subdomain interface have been recently examined and demonstrate varied degrees of tolerance for substitution (Spott et al. 2000).
An extremely interesting demonstration of plasticity in the LacI interface became apparent when attempts were made to use the monomeric nature of Y282D for other in vitro studies. Strikingly, either of two mutations at amino acid 84 (K84L or K84A) can compensate the Y282D dimerization defect, restoring assembly (Fig. 2 ▶; Chang et al. 1993). Residues 282 and 84 are spatially separated; in fact, they are located on different core subdomains (Fig. 2 ▶; Nichols et al. 1993; Friedman et al. 1995; Lewis et al. 1996; Bell and Lewis 2000). Therefore, this compensation is not due to rearrangements around the primary Y282D mutation. Instead, these K84 mutations must compensate the disruption caused by Y282D through globally strengthening the interface. In fact, both mutations at 84 in a wild-type background are extraordinarily resistant to chemical denaturation (Nichols and Matthews 1997). Hydrophobic mutations at position 84 also cause significant increases in the thermal stability of LacI (Gerk et al. 2000).
We hypothesized that other combinations of residues might compensate the dimerization defect of Y282D, either by increasing the global stability of dimer assembly or through local rearrangements around position 282. Designed mutations provide one avenue for exploring this issue, but such efforts are limited by our incomplete understanding of protein structures. Instead, we used the link between LacI assembly and function, which allows phenotypic screening for DNA binding of randomly mutated LacI. Because a dimer is the minimal unit for binding DNA, the mutation Y282D also destroys DNA binding. When K84L/A restores assembly to Y282D, DNA binding is also recovered. Any other second-site mutations generated through random mutagenesis that restore Y282D assembly should also restore DNA binding. This technique does not rely on structural assumptions and allows rapid screening of many candidates. In fact, the number of mutations, 22, that compensate the Y282D defect is surprisingly large, and many of the sites would not have been chosen for study by conventional techniques. We have examined these mutations in the context of the LacI structure and deduce that they restore effective assembly through multiple mechanisms.
Results and discussion
Comparison of repressor and periplasmic binding proteins
To explore the question of which residues are necessary for repressor dimerization, structures of the repressor core domains and several periplasmic binding proteins were used to create a structure-based alignment (Fig. 3 ▶; DALI: Holm and Sander 1996). For the repressor proteins, residues that have either side chain or backbone atoms within 3.5 Å of the partner monomer or hydrophobic contacts within 4.5 Å have been mapped onto this alignment. Interface differences between unliganded proteins, which have "open" structures, and those of the "closed" ligand-bound conformations are also noted. Additional water-mediated interactions (at least one atom from each monomer within 3.5 Å of the same water molecule) are listed in Table 1. In general, the latter also make direct contacts and are included in Figure 3 ▶, although water molecules mediate additional interactions for TreR 95 to 99 and PurR 284 to 284, 284 to 252, and 285/286 to 260.
Fig. 3.


Structure-based alignment of repressor and periplasmic binding proteins. Structure-based alignments were made by using the Dali server at the EMBL (http://www.emblheidelberg.de/Services/index.html) between the liganded "closed" structure of LacI (1lbh; Lewis et al. 1996) and the following proteins: PurR (1wet; Schumacher et al. 1994), TreR (1byk; Hars et al. 1998), RBP (2dri; Mowbray and Cole 1992), ABP (1abe; Newcomer et al. 1981; Quiocho and Vyas 1984), GBP (1glg; Vyas et al. 1988), and ALBP (1rpj; Chaudhuri et al. 1999). Gaps in the sequence of one protein relative to another are indicated with dashes. Dots are used to indicate loops "inserted" on ABP (41–66 and 239–249) that are too long to include on this figure or to indicate where sequence is not shown for simplicity (e.g., LacI 286–325). For the repressor proteins, residues with side chain or backbone contacts within 3.5 Å or hydrophobic contacts within 4.5 Å of the partner monomer are noted. Interactions of the liganded (closed) structure are represented with black text on a gray box; those of the unliganded (open) structure are indicated with white text on a gray box; and those present in both structures are shown as white text on a black box. The open structures used for LacI and PurR are 1efa and 1dbq, respectively (Schumacher et al. 1995; Bell and Lewis 2000). An open structure is not available for TreR. Two structures of closed, inducer-bound LacI are available, 1tlf and 1lbh (Friedman et al. 1995; Lewis et al. 1996). Contacts from both structures are included on this map. (A) Core-to-core contacts in the core N- and C-subdomains. Sites involved in the interfaces of all three repressors are indicated with Roman numerals I–X for the N-subdomain and I–VIII for the C-subdomain. (B) Monomer–monomer contacts between the HTH and core domains for LacI (open) and PurR (closed) are indicated, as well as LacI (closed) contacts between the core C-subdomain and the LHR. The latter are not available for the DNA-bound (open) structure of the protein.
Table 1.
Water-mediated monomer–monomer contacts
| Monomer A | Water | Monomer B | |
| LacI + DNA | 84,96 | 12 | 96 |
| (open, lefa) | 222 | 57 | 278 |
| (Bell and Lewis 2000) | 283 | 60 | 282,283 |
| Crystallographic waters are not available for IPTG-bound LacI | |||
| PurR unbound | 60(atom1) | 110 | 110 |
| (open, 1 dbq) | 82 | 59 | 69, 82 |
| (Schumacher et al. 1995) | 284 | 5 | 284 |
| 252 | 15 | 282, 284 | |
| 284 | 224 | 284 | |
| 260 | 18 | 285, 286 | |
| PurR + guanine + DNA | 260 | 13 | 286, 329 |
| (closed, 1 wet) | 286, 329 | 19 | 260 |
| (Schumacher et al. 1994) | |||
| TreR + Tre6P | 62 | 113 | 113, 117 |
| (closed, 1 byk) | 113, 117 | 13 | 62 |
| (Hars et al. 1998) | 99 | 7 | 89, 95 |
| 89, 95 | 107 | 99 | |
| 89 | 18 | 99 | |
| 99 | 118 | 89 | |
Numbers for monomers A and B correspond to the amino acid positions of residues involved in the water-mediated contacts. Numbers for waters correspond with those in their respective pdb files.
Core N-subdomain
Of the N-subdomain sites that directly contact their partner monomers, 10 are structurally homologous for all three repressors; these positions are indicated with Roman numerals I–X in Figure 3A ▶. However, no discrete pattern emerges from their amino acid identities that can delineate between oligomeric repressors and monomeric binding proteins. None of these sites have identical primary sequences for all three repressors. Amino acid similarity is only well-conserved as hydrophobic residues at sites VII and VIII, but the monomeric binding proteins also have hydrophobic side chains at these positions. In fact, an overall comparison for the N-core subdomain of RBP and the repressor proteins shows no significant deviation at any site. ABP does have a potentially disruptive proline at site II and a lysine at V and VIII. GBP has very low correlation at positions I and V, whereas ALBP is disparate at I, V, VII, and IX. However, given the low similarity between the three repressors, these differences are not remarkable. Sequence examination of this region does not reveal firm guidelines for oligomerization.
Since the primary sequences of these proteins demonstrate no striking patterns, we extended the analysis by looking for conserved interactions between pairs of sites. Contact maps were created for each interface to show the interactions that occur in the open and closed forms of each protein (Fig. 4 ▶). The open (ligand-free) structures of LacI and PurR have only one conserved interaction, between position III of monomer A and LacI residue 81 or PurR 79 on monomer B (Fig. 4 ▶A,B, dashed line). (The open structure of TreR is not available.) Four additional interactions create similar patterns on the contact maps of these two proteins (Fig. 4 ▶A,B, dotted lines): LacI has interactions between A:IV and B:VIII/B:IX that occur symmetrically from B to A. PurR has similar interactions using position V instead of IV. Otherwise, the interactions between unbound monomers produce strikingly different patterns. This variance is probably related to two issues. First, the open states of the two proteins differ the most at the core N-subdomain, both in their final positions and in their axes of rotation describing the motion that occurs on ligand-binding/release (Mowbray and Björkman 1999). Second, the two proteins have transposed functional modes. The open structure of LacI binds DNA, whereas PurR can only bind DNA in the closed complex with corepressor. In that light, both DNA-free forms of the proteins have interactions from A:VII to B:X and A:X to B:X. Further analysis of the N-terminal interface in the open, DNA-bound state must await new structural information from other repressors.
Fig. 4.


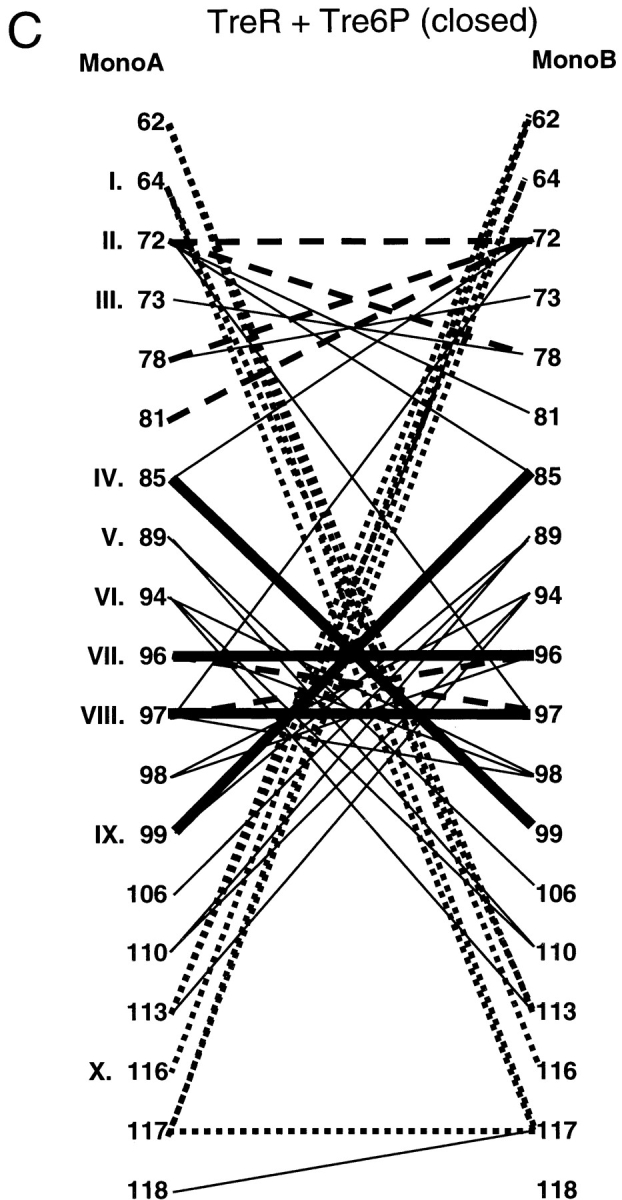



Interface contact maps. Specific interactions of the three repressors are mapped for their core N- and C-interfaces. Proteins and structures used are the same as those used in Figure 3 ▶. Amino acids common to the interfaces of all three proteins are indicated with Roman numerals that correspond to Figure 3 ▶. For LacI and PurR, the left side of the diagram represents the interface of the unliganded open structure, and the right side of the diagram is of the liganded, closed structure. Thick, solid lines represent interactions that are conserved between all three repressors. Dashed lines indicate interactions in common between any two repressors, and the dotted lines depict interactions that have significant similarities between any two proteins. Interactions unique to one repressor are symbolized with thin black lines. These contact maps do not include the cross-subdomain interactions between LacI 74 and 278 and PurR 70 and 278.
Even though their tertiary structures are more similar (Mowbray and Björkman 1999), the interaction maps of the closed core N-subdomains do not indicate many more common patterns. Only four interactions are conserved between the three repressors: A:VIII to B:IV, A:IV to B:VIII, VII to VII, and VIII to VIII (Fig. 4 ▶A–C, thick lines). Of these, position IV is conspicuous because several mutations of LacI 84 are known to have profound affects on interface strength (Chang et al. 1993; Nichols and Matthews 1997; Gerk et al. 2000). Some interactions are conserved between pairs of repressors, although no two proteins are more similar than the third (Fig. 4 ▶A–C, dashed lines). A few more similar but nonidentical patterns are indicated with dotted lines in Figure 4 ▶A–C. Therefore, the original sequence-based observation that the N-subdomain yields few strict rules of oligomerization appears to hold on structural analysis.
Core C-subdomain
Examination of the core C-subdomain does indicate a region that may be critical for assembly. This subdomain provides an unmoving anchor for the allosteric conformational changes of LacI and PurR (Schumacher et al. 1994, 1995; Friedman et al. 1995; Lewis et al. 1996; Mowbray and Björkman 1999; Bell and Lewis 2000; Matthews et al. 2000). Although the C-subdomain has fewer amino acids involved in the interface than does the N-subdomain, a greater percentage of sites are structurally conserved. These are labeled with Roman numerals I–VIII in Figure 3A ▶. Of these, positions I, II, V, and VI show little sequence similarity between all three repressors. In contrast, sites III, IV, VII, and VIII are all very hydrophobic. For the periplasmic binding proteins, this hydrophobicity is conserved at III and IV (with the exception of GBP III). However, the two protein families are not at all similar around VII and VIII. In fact, the primary sequences of the periplasmic binding proteins are so different from those of the repressors that their structural equivalence could not be inferred for a previous sequence-based alignment (Nichols et al. 1993). Differences arise not only from the primary sequences between the two groups at VII and VIII, but also from the insertions subsequent to position VIII that occur in all four periplasmic binding proteins examined (Fig. 3A ▶). For ABP, the six amino acids inserted in this region do not align with any other residues and, for simplicity, are indicated with dots.
Primary sequence mutations at site VIII had previously implicated this region in dimer assembly. Monomeric LacI Y282D was one of the earliest characterized mutations of this repressor (Schmitz et al. 1976; Daly and Matthews 1986; Chakerian and Matthews 1991; Chen and Matthews 1992b). In light of the known assembly states of LacI Y282 mutants, the nature of residues for the other aligned proteins is very intriguing. The PurR residue VIII is an F and TreR has an L; both substitutions are precisely those tolerated by LacI (Chakerian and Matthews 1991). Both PurR and TreR also have hydrophobic amino acids at position VII, and LacI C281 also has hydrophobic character (Fig. 3A ▶). None of the periplasmic binding proteins has hydrophobic residues at VII, although the S in ABP might be considered similar to the C of LacI. However, ABP has E at position VIII, where placing a D is fatal to LacI assembly. The VII–VIII regions for RBP and ALBP are also highly charged. Although the GBP sequence in the VII–VIII region is hydrophobic, this monomeric protein has an additional charged residue inserted on the loop subsequent to VIII. Interestingly, residue 289 of the LacI/GalR family member cytidine repressor also aligns with the site immediately following position VIII (Weickert and Adhya 1992). Substitution of a charged residue in the mutation C289R abrogates assembly of this normally oligomeric repressor (Barbier and Short 1993).
Contact maps of the core C-subdomain are also very striking (Fig. 4 ▶D–F). Most of the interactions are conserved among all three repressors (Fig. 4 ▶d–f, thick lines). The pattern is most noticeable for TreR, which has only four unique interactions (Fig. 4F ▶, thin lines). The open and closed C-interfaces of LacI and PurR exhibit few differences, consistent with previous observations that this subdomain is not affected by the allosteric conformational change (Fig. 4 ▶D,E; Schumacher et al. 1994, 1995; Friedman et al. 1995; Lewis et al. 1996; Mowbray and Björkman 1999; Bell and Lewis 2000; Matthews et al. 2000). The five interactions of residue VII are particularly striking and appear to provide the key stabilizing interactions of this interface. Structurally, the side chain of this residue provides the "ball" for a hydrophobic "socket" of its partners (Fig. 5 ▶)
Fig. 5.
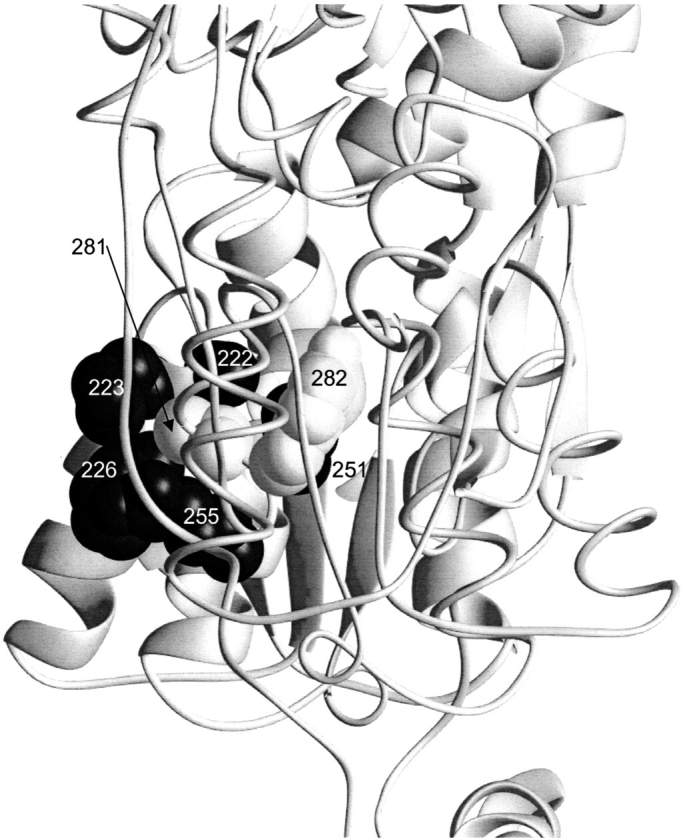
Interactions of C281. In this view, the dimeric unit of LacI shown in Figure 2 ▶ is turned 90° out of the plane of the paper (1lbh; Lewis et al. 1996). The sidechains for C281 and Y282 on monomer A are indicated in gray (backbone interactions for these positions are not apparent on this figure). The rest of monomer A is depicted as a thin ribbon for clarity. Positions 222, 223, 226, 251, and 255 on monomer B are shown as black balls. Complementary interactions between B:281/282 and A:222, 223, 226, 251, and 255 are not shown.
However, this site also provides an excellent example of why structural data alone are not sufficient for deciphering the code of protein structure. Both in vivo and in vitro studies demonstrate that position VII of LacI nonetheless tolerates a wide variety of substitutions. LacI C281 mutation to S, A, F, I, or M results in biophysical characteristics similar to wild-type protein (Chakerian and Matthews 1991). Phenotypic analysis of C281 mutations to L, H, Y, F, A, S, Q, R, or E demonstrates that these mutants all maintain their repression function (Suckow et al. 1996), which would be destroyed if assembly were abolished. Only C281 altered to P, K, and G diminished repression (Suckow et al. 1996). C281 has also been the target of extensive chemical modification (Daly et al. 1986). Low reaction rates indicate that C281 is shielded from solvent and is modified by only extremely reactive compounds, generally at high concentrations of reactants. However, the function of the resulting protein is only moderately affected, and the modified interface must still be intact (Daly et al. 1986). Thus, LacI position VII demonstrates plasticity even beyond the options provided by nature.
In contrast, LacI mutations at position VIII, which makes only two contacts in the interface (Fig. 4D ▶), are poorly tolerated. Chakerian and Matthews (1991) demonstrated that only F and L are tolerated; A, S, and E abolish oligomerization. Phenotypic analysis yielded similar results: L and F maintained wild-type phenotypes whereas P, H, C, A, G, S, N, K, R, or E substitutions abolished repression function (Suckow et al. 1996). Our current hypothesis is that the differing mutational sensitivities arise from the proximity of each site to the surface of the interface. Site VII is on the edge, but VIII is buried in the middle and therefore has stricter packing constraints.
Phenotypic screen
Despite mutational sensitivity at site VIII, both assembly and function of the LacI Y282D mutation are restored by a second-site mutation at K84. To determine how many second-site mutations can revert the phenotype of Y282D and thereby explore plasticity of quaternary structure, we developed tools to screen the function of a large number of randomly mutated proteins. The phenotypic screen presented in Figure 6 ▶ required several stages of development but evolved into a facile and effective tool with reproducible results even in the hands of novice researchers. The detailed rationale of the screen is as follows. Wild-type LacI (WT) is expressed from the repressor plasmid and binds to its operator site on the pZCam reporter plasmid. This binding represses transcription of β-galactosidase, and colonies remain white in the presence of the indicator Xgal. If the Y282D mutation is present on the repressor plasmid, the resulting LacI protein is monomeric and cannot bind DNA, allowing β-galactosidase transcription and resulting in turquoise blue colonies in the presence of Xgal. If a second mutation restores dimerization of Y282D protein, so that resulting protein can again bind operator DNA and inhibit β-galactosidase transcription, colonies remain white in the presence of Xgal. If the doubly mutated protein exists in equilibrium between monomer and dimer, it will not repress as efficiently as wild-type LacI and may produce light blue colonies. This last feature confers an advantage to the blue/white screen over alternative toxicity selection approaches, which could not detect intermediates. A further benefit of this screen is that the high ratio of repressor protein to reporter plasmid can allow detection of double mutants that may form only weakly associated oligomers.
Fig. 6.

Schematic of phenotypic screen for Y282D revertants.
Results of the genetic screen of >30,000 colonies are presented in Table 2. The mutations that compensate the dimerization defect of Y282D produced either white or very light blue colonies. A large percent of light blue colonies identified in the screen were found to be false positives (Y282D with no further mutations), as well as a few of the white colonies. Although this result raises the possibility that some of the mutations detected by the screen do not actually affect LacI assembly or function, several lines of evidence support the position that they do compensate the Y282D defect. The most substantive evidence is structural analysis; sites are easily assigned to one of three classes (see below). An especially interesting group contains sequential sites: 149, 150, and 151. Furthermore, seven revertants were each detected more than once (Table 2), five of which (M42I, H74R, N246S, T276I, and S354F) were isolated from Y282D plasmids mutated in separate experiments. Finally, four of the sites (133, 223, 274, and 276) had multiple mutations (Table 2).
Table 2.
Results from two blue/white phenotypic screens
| Number of colonies screened | ∼31,000 |
| Positives after two rounds of screening | 129 |
| Number with DNA sequenced | 87 |
| Number of D282Y (wild-type revertant) | 17 |
| Number false positives (no additional mutation) | 36 |
| Mutations identified (frequency) | M42I (3) |
| H74R (3) | |
| M98I (2) | |
| A133: T(1); V (1) | |
| D149N (1) | |
| V150I (1) | |
| S151P (1) | |
| S191F (2) | |
| M223: I(1); TG (1) | |
| M232I (1) | |
| N246S (2) | |
| Q248R (1) | |
| D274: N (1); G (1) | |
| T276: A (1); I (2) | |
| P284S (1) | |
| L296M (1) | |
| V321I (1) | |
| S354F (5) |
Since all of the reversion mutations resulted from a single base pair change, we found their number to be surprisingly large. Furthermore, the K84 mutations known to compensate Y282D (Chang et al. 1993) were not obtained, because both require changing more than one base pair, implying that mutagenic saturation has not yet been reached. Although we attempted higher frequency mutation by passaging singly mutated plasmids back through XL1-Red cells, these plasmids were corrupted beyond use (data not shown). Further efforts using PCR-based methods are being pursued to exhaustively identify second-site revertants for the Y282D phenotype.
One other result that bears note is that some of the wild-type revertants (D282Y) included mutations at one or more of the three cysteine residues (C107, C140, and C281; see Table 2). These were the only revertants containing more than one mutation per LacI gene. We find these results very intriguing in light of challenges encountered when altering these cysteines in other projects (Wycuff 1999).
Sites of reversion
The mutations that compensate for the dimerization defect caused by Y282D are listed in Table 2. These sites were mapped onto the X-ray crystallographic structures for LacI (Friedman et al. 1995; Lewis et al. 1996; Bell and Lewis 2000). The sites of mutations cluster into three groups: (1) residues where changes might globally strengthen the oligomerization interfaces in a manner analogous to K84L; (2) side chains positioned so that mutations might elicit local structural rearrangement and compensate the effects of Y282D; and (3) sites that may either affect long-range structural changes, shifting LacI to a state with high affinity for DNA, or increase the DNA binding affinity of a species for which it was not previously detectable.
Global stabilization of oligomerization interfaces: H74R, M98I, M223I/T, M232I, Q248R, and S354F
The original mutations known to compensate Y282D oligomerization (K84L and K84A) appear to function through strengthening the monomer–monomer interface (Nichols and Matthews 1997; Gerk et al. 2000). Therefore, the fact that the first two mutants identified by this study, H74R and M98I, are also located on the core N-subdomain in the monomer–monomer interface was very satisfying (Fig. 2 ▶). In fact, the contacts between M98 and K84 are two of the four interactions conserved in the closed core N-interface for all three repressors (Fig. 4 ▶A–C). M98I may therefore function by forming a more extensive hydrophobic patch at the interface. Similarly, M223 is involved in a cross-monomer interaction of the core C-subdomain with S280 (Lewis et al. 1996). The LacI reversion mutation M223I/T may also strengthen the hydrophobicity of this contact (Fig. 7 ▶).
Fig. 7.

Local mutants. This structure depicts the core C-subdomain interface for one of the two dimers of the DNA-bound structure of LacI (1efa; Bell and Lewis 2000). Positions of mutations that revert the phenotype of Y282D (223, 246, 248, 274, 276, and 284) are indicated with spheres. D278, which does not form an ion pair with H74 in this conformation, is indicated with the smaller balls and sticks.
In contrast, an ionic interaction occurs between H74 and D278 when wild-type LacI binds inducer (Friedman et al. 1995; Lewis et al 1996). This interaction is the sole contact between the core N-subdomain of one monomer and the core C-subdomain of its partner monomer and is not present when LacI binds DNA (Fig. 2 ▶; Lewis et al. 1996; Bell and Lewis 2000). In fact, the cross-monomer partner for D278 in the "open" DNA-bound conformation is Q248 (Fig. 7 ▶; Bell and Lewis 2000). Interestingly, both H74R and Q248R compensate the Y282D lesion. These structural observations led to an initial hypothesis that the H74/D278 ion pair is essential for allosteric communication. However, Barry and Matthews (1999) found that, although mutations at position 74 do affect allosteric communication, the ionic bond is not required. Because arginine should form a stronger ion pair than histidine, the original hypothesis would predict that H74R should strengthen the inducer-bound form of LacI, which cannot bind DNA with high affinity. Therefore, the observation that the H74R mutation allows Y282D to bind operator DNA supports the conclusions of Barry and Matthews (1999). The stronger base (arginine) in H74R (or Q248R) may compensate for the additional negative charge of the Y282D. Interestingly, E70 of PurR makes a comparable ionic bond with PurR R278, and an E70A mutation (which removes a negative charge) results in a shift to the closed conformation, which has high affinity for DNA (Lu et al. 1998).
The locations of two other reversion mutations, M232I and S354F, have surprising implications (Fig. 2 ▶). S354 is in the LHR tetramerization region of LacI (Fig. 2 ▶). In wild-type LacI, residues 351 and 355 of one monomer contact the core domain of its partner monomer (Fig. 3 ▶); this interaction is asymmetric in that it occurs only once per dimer. Therefore, we speculate that the S354F mutation may provide an additional LHR-to-core interaction, thus stabilizing the assembled form of the protein. In contrast, M232 is not near any known or potential monomer–monomer interfaces. In fact, residue 232 is on a loop that juts into the space between dimers and may form a second dimer–dimer interface (in addition to the LHR; not shown). We speculate that M232I again provides a tighter hydrophobic contact, stabilizes the tetramer, and by Le Chatelier's principle, strengthens the dimers. This effect on the linked assembly steps is opposite to that observed for the L251A mutation (Dong et al. 1999).
Local structural effects: M223I/T, N246S, Q248R, D274N/G, T276A/I, and P284S
Another simple mechanism for compensating the Y282D mutation is through local adjustments in the structure that accommodate the tyrosine to aspartate change. Indeed, nine mutations—M223I/T, N246S, Q248R, D274N/G, T276A/I, and P284S—all occur in the immediate area of the original Y282D mutation (Fig. 7 ▶), and we postulate that they function in this manner. M223I/T and Q248R also participate in the monomer–monomer interface (see earlier). In particular, one means by which Q248R and D274N/G might compensate Y282D is through reducing excessive negative charge created by the aspartate. Several of these "local" sites are directly involved in wild-type LacI inducer binding (Friedman et al. 1995; Lewis et al. 1996). Interestingly, the phenotypes of E. coli expressing genes encoding some of these compensating mutations in a wild-type (Y282) background have been examined by the Miller lab and found to alter allosteric response to inducer (Kleina and Miller 1990; Suckow et al. 1996).
Relevant details for each site follow. N246 directly contacts inducer IPTG (Friedman et al. 1995; Lewis et al. 1996), and the mutation N246S is classified as having weak inducer sensitivity by Kleina and Miller (1990). These authors likewise report that Q248R in a wild-type background is insensitive to inducer (Kleina and Miller 1990). D274 also contacts IPTG (Friedman et al. 1995; Lewis et al. 1996), and D274N/wild type cannot bind inducer in vitro (Chang et al. 1994). D274G also has an inducer-insensitive phenotype, as does T276A (Suckow et al. , 1996). Experiments to determine the effects of these mutations within the context of Y282D on inducer binding and allosteric response promise interesting results. However, instead of further phenotypic analysis or approximate assays of β-galactosidase activity, we have opted to begin purification and biophysical characterization of the double revertant/Y282D mutants. This decision was based on our experience with K84L, which has an inducer-insensitive phenotype (Kleina and Miller 1990; Suckow et al. 1996) but wild-type thermodynamics of IPTG binding (Chang et al. 1993). The discrepancy in the results from the two techniques is actually due to extraordinarily slow kinetics of inducer binding (Chang et al. 1993).
Several features of the mutation P284S warrant additional comment. This site is the only "local" residue not located near the inducer binding pocket and may deserve to be categorized separately (Fig. 7 ▶). In a wild-type background, P284S cannot bind DNA and is a heat-sensitive mutation (Suckow et al. 1996). Although this site is not in the monomer–monomer interface, it is directly adjacent to the 281–282 region postulated from the structural alignment (Figs. 3, 4 ▶ ▶) to be critical to dimerization. Perhaps this single mutation also disrupts assembly, whereas the two mutations P284S and Y282D are complementary, allowing assembly and DNA binding.
Putative allosteric mutants: M42I, A133T/V, D149N, V150I, S151P, S191F, L296M, and V321I
The locations of the remaining mutants identified as Y282D revertants are quite surprising (Figs. 8, 9 ▶ ▶). In fact, when A133V was first detected, we considered the possibility that it could be an artifact of the phenotypic screen until a cluster of revertant sites—A133T/V, D149N, V150I, S151P, and S191F—became apparent behind the inducer binding site on the face opposite the monomer–monomer interface (Fig. 8 ▶). In a wild-type background, both D149 and S191 contact inducer (Friedman et al. 1995; Lewis et al. 1996), and the S151P and S191F mutations cause an inducer-insensitive phenotype (Suckow et al. 1996). This region of the protein also appears to be intimately involved in the allosteric conformational change that occurs with inducer binding, in which the core N-subdomains move apart from each other while the core C-subdomains are fixed (Lewis et al. 1996; Bell and Lewis 2000; Matthews et al. 2000). A similar conformational change occurs in PurR, although in this case corepressor is required for assuming the DNA high-affinity conformation (Schumacher et al. 1994, 1995). Interestingly, the PurR mutation W147A shifts the protein to the conformation with high affinity for DNA, even in the absence of corepressor (Lu et al. 1998). Because the PurR residue 147 aligns with LacI 150 (Fig. 3 ▶), a similar mechanism may apply for this set of Y282D revertants, in particular the LacI V150I. Stabilization of the LacI DNA-bound form may facilitate detection of extremely low levels of assembled Y282D not perceptible by in vitro characterization.
Fig. 8.
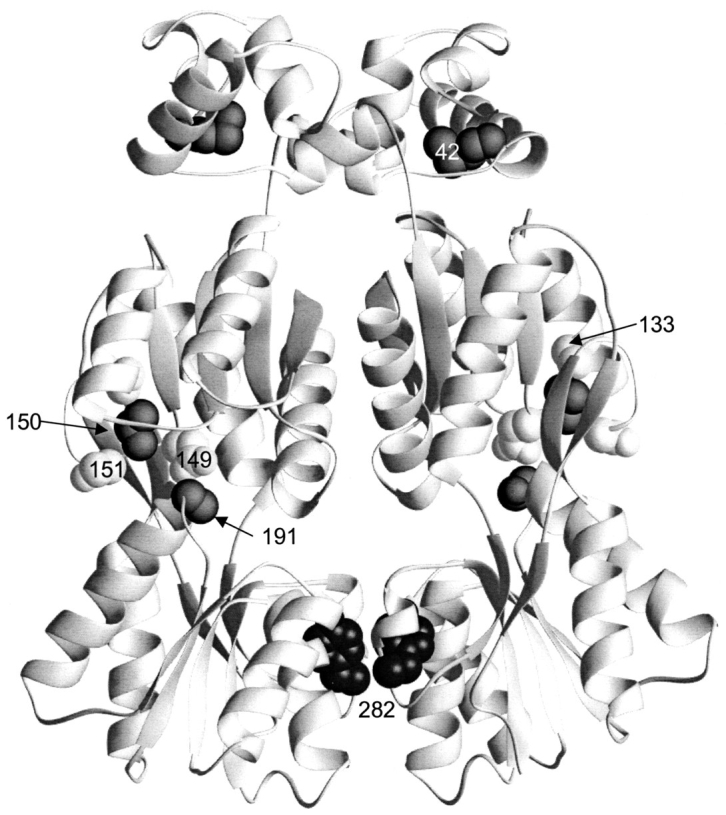
Allosteric or DNA-binding mutants. This structure depicts one of the two dimers of the DNA-bound structure of LacI (1efa). The LHR that mediates tetramerization is not present in this structure, because a dimeric mutant of LacI was used (Bell and Lewis 2000). Positions of mutations that revert the phenotype of Y282D (42, 133, 149, 150, 151, and 191) are indicated with spheres.
Fig. 9.

Strands connecting core N- and C-subdomains. This structure depicts one of the two dimers of the IPTG-bound structure of LacI (1lbh; Lewis et al. 1996). The N-terminal DNA binding domain is not resolved for this structure. The three strands that connect the two subdomains of the core are indicated with thick ribbons. The positions of 296 and 321, mutations of which revert the monomeric phenotype of Y282D, are indicated with spheres on these strands.
Although L296M and V321I do not adjoin the cluster around residue 150, they are each on one of the three strands that covalently link the core N- and C-subdomains (Fig. 9 ▶) and are affected by induction when the core N-subdomains rotate together. Therefore, we find it reasonable to extend the hypothesis of long-range conformational effect to these residues. As we proceed with in vitro characterization, this group of intriguing mutants will be of particular interest.
Finally, we include M42I in this category because it probably also affects DNA binding. M42 is in the DNA-binding domain (Fig. 8 ▶), and the recent high-resolution structure of DNA-bound dimeric LacI shows the M42 side chain pointing toward the C terminus of helix 2 (Chuprina et al. 1993; Slijper et al. 1996; Bell and Lewis 2000). The M42I mutation may therefore stabilize the HTH DNA-binding domain, generating a protein with higher affinity for DNA binding. This mutation might allow detection of small populations of Y282D oligomer. Alternatively, the DNA binding affinity of M42I might be increased enough to detect binding of LacI monomer, which normally binds with an affinity 10,000-fold less than those of assembled species.
Conclusion
The ability of many different amino acid sequences to assume similar folds demonstrates the plasticity of secondary and tertiary structures. The structurally homologous families of LacI/GalR repressors and periplasmic binding proteins provide good examples of this behavior (von Wilcken-Bergmann and Müller-Hill 1982; Müller-Hill 1983; Weickert and Adhya 1992). However, few studies have explored whether the same principles apply to quaternary structure.
Structural alignments of monomeric binding proteins and oligomeric repressors, as well as contact maps of the latter, indicate that the core N-subdomain does not comprise many of the critical dimerization interactions. This arrangement is highly amenable to the concept of quaternary plasticity. Exceptions to these statements are that the positions equivalent to LacI 84, 95, 96, and 98 participate in conserved interactions of the closed conformations. Interestingly, mutations at 84 have previously been identified to have significant effects on interface strength (Chang et al. 1993), and this paper reports that M98I may have similar properties. The core C-subdomain interface, in contrast, is composed of many conserved interactions. Although half of the sites that comprise this interface do not demonstrate primary sequence conservation, both sequence and structural analyses indicate the sites corresponding to LacI 281 and 282 are especially important. From these observations, one might predict that any mutations at these sites would be detrimental to oligomeric assembly. Although this hypothesis is consistent with experimental data for 282, mutations at 281 have little or no effect on LacI oligomerization. This result for 281 provides yet another example of quaternary plasticity in repressor assembly.
The monomeric Y282D mutation of LacI has provided an advantageous context for further examining the issue of plasticity in assembly. Random mutagenesis and phenotypic screening for Y282D revertants have identified a surprisingly broad range of sites that appear to represent several different mechanisms for correcting the oligomerization defect. Many of these sites would have been unlikely targets of "rational" experimental design. The diverse regions identified in this study indicate that determinants of oligomeric assembly and DNA affinity are remarkably flexible. Detailed biochemical and biophysical characterization of these mutant proteins will be required to identify the precise mechanisms by which they mitigate the dramatic effect of Y282D on assembly and to determine any functional consequences that result from changing these amino acids. This information will accrue from measurements of the thermodynamic parameters of assembly and ligand binding of the purified mutant proteins.
The mutagenesis experiments indicate that the current 360-amino acid scaffold for LacI tolerates a broad range of changes without abolishing the potential for assembly. The strength of the LacI interface comes from many small interactions, and the precise nature of the contributors is not stringent. Indeed, energetic loss at one position (e.g., Y282D) can be compensated by changing a variety of second sites. Furthermore, the primary sequences of the sites involved in the repressor interfaces are not significantly distinct from their analogous residues of the monomeric periplasmic binding proteins, except near C-subdomain positions VII/VIII. However, for LacI, position VII (281) tolerates mutagenesis, and a deleterious substitution at position VIII (282) is easily compensated with second-site mutations. Therefore, the binding proteins perhaps need only a few additional interactions to facilitate formation of quaternary structures similar to those of the repressors.
These two families have one other subtle but possibly critical difference that affects not only their primary but also their tertiary structures. The presence of additional amino acids subsequent to position VIII in the core C-subdomains of the monomeric periplasmic binding proteins, but not the oligomeric repressors, may abrogate oligomerization. This situation suggests several interesting experimental questions: Will addition of this loop to LacI abolish assembly? If so, can the monomer–monomer interface be restored by second-site mutations in a manner analogous to the Y282D revertants? Will elimination of this loop in the monomeric binding proteins allow their assembly? The answers to these questions will help define how closely the plasticity demonstrated for LacI quaternary structure is coupled to its underlying tertiary fold.
Materials and methods
Plasmids and random mutagenesis
The reporter plasmid pZCam was constructed by excising the LacZ gene for β-galactosidase and its lac operator from pCRII (Invitrogen) with the restriction enzyme AflIII. The resulting overhanging ends were filled in with Klenow fragment, and the product was inserted into the tetracycline resistance gene of pACYC184 (NEB) at the blunt ends of an EcoRV restriction site. pZCam retains the Cam resistance gene of pACYC184 and an origin of replication that is compatible with the ColE1 origin of the plasmids containing the various LacI mutations. The latter carry the β-lactamase gene for Amp resistance.
The wild-type LacI gene was on plasmid pLS1, which differs from previously reported pJC1 (Chen and Matthews 1992a) in that a silent mutation at amino acid 45 to make a SacI restriction site was added by using Stratagene's Chameleon Double-Stranded Site Directed Mutagenesis protocol, the mutagenic oligo 5′-GTTGG GAATGTAATTGAGCTCCGCCATCGC-3′, and the selection oligo 5′-GATCCGTCGACCTCGAGCCAAGCTTG-3′. The latter changes the PstI site of pJC1 to an XhoI site on pLS1.
The Y282D mutation of LacI was on the plasmid pAC1 (Chakerian and Matthews 1991). The use of this plasmid may have been fortuitous in detecting the large number of revertants. Different LacI plasmids have various numbers of DNA binding sites, and adding even one extra site can change the detection level of the screen (results not shown). Y282D plasmid was randomly mutated by passaging the DNA through XL1-Red cells (Epicurian Coli, Stratagene). XL1-Red cells are deficient in DNA repair mechanisms, and errors in replication occur approximately once per kilobasepair. Because the LacI gene is 1080 basepairs long, mutagenesis in this system averaged one base per gene.
Blue–White phenotypic screen
Many changes in the LacI DNA binding function are readily observed by using a phenotypic screen (Kleina and Miller 1990; Markiewicz et al. 1994; Suckow et al. 1996). However, previously constructed screens rely primarily on nonsense suppression at known amino acid positions and incorporate the LacI mutations into the episome, which fixes the repressor:reporter gene ratio. This work required a screen that allowed random mutagenesis, variability in the repressor:reporter ratio (for future applications), easy purification of modified DNA for sequencing, and the facility to transform the DNA into various E. coli strains (again, for future applications). These constraints are all met by keeping the LacI gene on a plasmid such as pAC1 or pLS1.
The ideal reporter gene would have been incorporated into an E. coli strain that had a deleted LacI gene. However, we could locate only interrupted-LacI/LacZ combinations, which we believed potentiated the hazard of homologous recombination. Therefore, we chose a system in which the LacZ reporter gene was carried on the cotransformable plasmid (pZCam, see earlier). We first transformed pZCam into E. coli DH5α cells (GIBCO BRL), then created new competent cells designated DZCam (using CaCl2), and then transformed the various LacI plasmids.
Randomly mutated Y282D DNA was transformed into competent DZCam cells, which were plated on Cam/Amp LB agar plates and grown at 37°C overnight. Control plates contained bacterial colonies expressing wild-type and Y282D LacI. All colonies were then sprayed with 40 mg/mL Xgal in dimethylformamide by using a TLC reagent sprayer and incubated at 4°C in the dark. After ∼7 h, colonies were again sprayed with Xgal and incubated ∼16 h under the same conditions. After this process, Y282D control colonies were dark blue, whereas colonies expressing wild-type LacI remained white (Fig. 6 ▶). The latter was always verified, because even wild-type LacI will allow expression of a small amount of β-galactosidase over extended periods of time, causing the colonies to become light blue. Any colonies transformed with mutated DNA that were white or lighter blue than the Y282D control were streaked on Cam/Amp LB plates and incubated at 37°C overnight. Xgal was applied in the same manner to verify the original phenotype.
Sequencing positive colonies
If the streaked colonies remained white or lighter blue than Y282D control colonies, they were transferred to 5 mL 2XYT or LB liquid media (with Amp and Cam) and grown overnight. Plasmid DNA was purified from these cells by using the Wizard Plus Miniprep DNA Purification Kit (Promega) and sent for full sequencing to either Genosys Biotechnologies, Inc. or Lone Star Labs, Inc. Although this DNA was a mixture of LacI plasmid and reporter plasmid pZCam, the latter did not interfere with sequencing because of its low copy number. After desirable mutations were identified, the LacI plasmid was separated from pZCam by using the following scheme. The plasmid mixture was incubated with the SacII restriction enzyme, which digests only the pZCam plasmid. The mixture was then transformed into DH5α cells and plated on LB/Amp agarose plates. After an overnight, 37°C incubation, new colonies were gridded onto both Amp/LB and Cam/LB agarose plates. DNA was purified from colonies that grew only on the Amp plate (generally 100% of the colonies gridded).
Acknowledgments
We thank Duane A. Beasley for assistance in constructing pZCam and Caroline Wu, Eric T. Lee, and Atul Maheshwari for assistance in screening colonies. Simon Sims and Genosys Inc. graciously provided DNA sequencing. L.S.K. was supported by a training fellowship from the W. M. Keck Center for Computational Biology (National Library of Medicine Grant LM07093). This work was supported by grants to K.S.M. from the National Institutes of Health (GM22441) and the Robert A. Welch Foundation (C-576).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
LacI, lactose repressor protein
PurR, purine repressor protein
TreR, trehalose repressor protein
GBP, glucose/galactose binding protein
RBP, ribose binding protein
ABP, DL-arabinose binding protein
ALBP, D-allose binding protein
IPTG, isopropyl-1-thio-β,D-galactoside
HTH, helix-turn-helix DNA binding domain
LHR, leucine heptad repeat tetramerization domain
Cam, chloramphenicol
Amp, ampicillin
Xgal, 5-bromo-4-chloro-3-indolyl-β,D-galactoside.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.35801.
Footnotes
The Escherichia coli LacI has long served as a prototype for proteins that negatively control transcription. In vivo, LacI binds tightly to a specific operator DNA site, preventing transcription of downstream lactose metabolic genes. If lactose becomes the predominant energy source, the bacteria first produce the metabolite allolactose, which binds to LacI and induces a conformational switch to a protein structure with only nonspecific DNA-binding affinity. The large excess of genomic bacterial DNA effectively competes to bind inducer–repressor complex. LacI vacates the operator site, thus allowing transcription of the downstream metabolic genes. The lac system is used to control many recombinant protein expression systems, substituting the gratuitous inducer IPTG, because it is not metabolized by E. coli (for review see Matthews and Nichols 1998).
References
- Alberti, S., Oehler, S., von Wilcken-Bergmann, B., Krämer, H., and Müller-Hill, B. 1991. Dimer-to-tetramer assembly of Lac repressor involves a leucine heptad repeat. The New Biologist 3 57–62. [PubMed] [Google Scholar]
- Alberti, S., Oehler, S., von Wilcken-Bergmann, B., and Müller-Hill, B. 1993. Genetic analysis of the leucine heptad repeats of Lac repressor: Evidence for a 4-helical bundle. EMBO J. 12 3227–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Barbier, C.S. and Short, S.A. 1993. Characterization of cytR mutations that influence oligomerization of mutant repressor subunits. J. Bacteriol. 175 4625–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry, J.K. and Matthews, K.S. 1999. Substitutions at histidine 74 and aspartate 278 alter ligand binding and allostery in lactose repressor protein. Biochemistry 38 3579–3590. [DOI] [PubMed] [Google Scholar]
- Bell, C.E. and Lewis, M. 2000. A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol. 7 209–214. [DOI] [PubMed] [Google Scholar]
- Björkman, A.J. and Mowbray, S.L. 1998. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol. 279 651–664. [DOI] [PubMed] [Google Scholar]
- Chakerian, A.E. and Matthews, K.S. 1991. Characterization of mutations in oligomerization domain of lac repressor protien. J. Biol. Chem. 266 22206–22214. [PubMed] [Google Scholar]
- Chakerian, A.E., Tesmer, V.M., Manly, S.P., Brackett, J.K., Lynch, M.J., Hoh, J.T., and Matthews, K.S. 1991. Evidence for leucine zipper motif in lactose repressor protein. J. Biol. Chem. 266 1371–1374. [PubMed] [Google Scholar]
- Chang, W.I., Olson, J.S., and Matthews, K.S. 1993. Lysine 84 is at the subunit interface of lac repressor protein. J. Biol. Chem. 268 17613–17622. [PubMed] [Google Scholar]
- Chang, W.I., Barrera, P., and Matthews, K.S. 1994. Identification and characterization of aspartate residues that play key roles in the allosteric regulation of a transcription factor: Aspartate 274 is essential for inducer binding in lac repressor. Biochemistry 33 3607–3616. [DOI] [PubMed] [Google Scholar]
- Chaudhuri, B.N., Ko, J., Park, C., Jones, T.A., and Mowbray, S.L. 1999. Structure of D-allose binding protein from Escherichia coli bound to D-allose at 1.8 Å resolution. J. Mol. Biol. 286 1519–1531. [DOI] [PubMed] [Google Scholar]
- Chen, J. and Matthews, K.S. 1992a. Deletion of lactose repressor carboxyl-terminal domain affects tetramer formation. J. Biol. Chem. 267 13843–13850. [PubMed] [Google Scholar]
- Chen, J. and Matthews, K.S. 1992b. T41 mutation in lac repressor is Tyr282 →Asp. Genes 111 145–146. [DOI] [PubMed] [Google Scholar]
- Chuprina, V.P., Rullmann, J.A.C., Lamerichs, R.M.J.N., van Boom, J.H., Boelens, R., and Kaptein, R. 1993. Structure of the complex of lac repressor headpiece and an 11 base-pair half-operator determined by nuclear magnetic resonance spectroscopy and restrained molecular dynamics. J. Mol. Biol. 234 446–462. [DOI] [PubMed] [Google Scholar]
- Daly, T.J. and Matthews, K.S. 1986. Characterization and modification of a monomeric mutant of the lactose repressor protein. Biochemistry 25 5474–5478. [DOI] [PubMed] [Google Scholar]
- Daly, T.J., Olson, J.S., and Matthews, K.S. 1986. Formation of mixed disulfide adducts at cysteine-281 of the lactose repressor protein affects operator and inducer binding parameters. Biochemistry 25 5468–5474. [DOI] [PubMed] [Google Scholar]
- Dong, F., Spott, S., Zimmermann, O., Kisters-Woike, B., Müller-Hill, B., and Barker, A. 1999. Dimerisation mutants of lac repressor. I. A monomeric mutant, L251A, that binds lac operator DNA as a dimer. J. Mol. Biol. 290 653–666. [DOI] [PubMed] [Google Scholar]
- Files, J.G. and Weber, K. 1976. Limited proteolytic digestion of lac repressor by trypsin: Chemical nature of the resulting trypsin-resistant core. J. Biol. Chem. 251 3386 – 3391. [PubMed] [Google Scholar]
- Friedman, A.M., Fischmann, T.O., and Steitz, T.A. 1995. Crystal structure of lac repressor core tetramer and its implications for DNA looping. Science 268 1721–1727. [DOI] [PubMed] [Google Scholar]
- Geisler, N. and Weber, K. 1977. Isolation of the amino-terminal fragment of lactose repressor necessary for DNA binding. Biochemistry 16 938–943. [DOI] [PubMed] [Google Scholar]
- Gerk, L.P., Leven, O., and Müller-Hill, B. 2000. Strengthening the dimerisation interface of lac repressor increases its thermostability by 40 deg C. J. Mol. Biol. 299 805–812. [DOI] [PubMed] [Google Scholar]
- Hars, U., Horlacher, R., Boos, W., Welte, W., and Diederichs, K. 1998. Crystal structure of the effector-binding domain of the trehalose-repressor of Escherichia coli, a member of the LacI family, in its complexes with inducer trehalose-6-phosphate and noninducer trehalose. Protein Sci. 7 2511–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1996. Mapping the protein universe. Science 273 595–560. [DOI] [PubMed] [Google Scholar]
- Jovin, T.M., Geisler, N., and Weber, K. 1977. Amino-terminal fragments of Escherichia coli lac repressor bind to DNA. Nature 269 668–672. [DOI] [PubMed] [Google Scholar]
- Khoury, A.M., Nick, H.S., and Lu, P. 1991. In vivo interaction of Escherichia coli lac repressor N-terminal fragments with the lac operator. J. Mol. Biol. 219 623–634. [DOI] [PubMed] [Google Scholar]
- Kleina, L.G. and Miller, J.H. 1990. Genetic studies of the lac repressor. XIII. Extensive amino acid replacements generated by the use of natural and synthetic nonsense suppressors. J. Mol. Biol. 212 295–318. [DOI] [PubMed] [Google Scholar]
- Lewis, M., Chang, G., Horton, N.C., Kercher, M.A., Pace, H.C., Schumacher, M.A., Brennan, R.G., and Lu, P. 1996. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science 271 1247–1254. [DOI] [PubMed] [Google Scholar]
- Lu, F., Brennan, R.G., and Zalkin, H. 1998. Escherichia coli purine repressor: Key residues for the allosteric transition between active and inactive conformations and for interdomain signaling. Biochemistry 37 15680–15690. [DOI] [PubMed] [Google Scholar]
- Markiewicz, P., Kleina, L.G., Cruz, C., Ehret, S., and Miller, J.H. 1994. Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as "spacers" which do not require a specific sequence. J. Mol. Biol. 240 421–434. [DOI] [PubMed] [Google Scholar]
- Matthews, K.S. and Nichols, J.C. 1998. Lactose repressor protein: Functional properties and structure. Prog. Nucl. Acid Res. Mol. Biol. 58 127–164. [DOI] [PubMed] [Google Scholar]
- Matthews, K.S., Falcon, C.M., and Swint-Kruse, L. 2000. Relieving repression. Nat. Struct. Biol. 7 184–187. [DOI] [PubMed] [Google Scholar]
- Mowbray, S.L. and Björkman, A.J. 1999. Conformational changes of ribose-binding protein and two related repressors are tailored to fit the functional need. J. Mol. Biol. 294 487–499. [DOI] [PubMed] [Google Scholar]
- Mowbray, S.L. and Cole, L.B. 1992. 1.7Å X-ray structure of the periplasmic ribose receptor from Escherichia coli. J. Mol. Biol. 177 155–175. [DOI] [PubMed] [Google Scholar]
- Müller-Hill, B. 1983. Sequence homology between Lac and Gal repressors and three sugar-binding periplasmic proteins. Nature 302 163–164. [DOI] [PubMed] [Google Scholar]
- Newcomer, M.E., Gilliland, G.L., and Quiocho, F.A. 1981. L-Arabinose-binding protein-sugar complex at 2.4Å resolution. J. Biol. Chem. 256 13213–13217. [PubMed] [Google Scholar]
- Nichols, J.C. and Matthews, K.S. 1997. Combinatorial mutations of lac repressor: Stability of monomer-monomer interface is increased by apolar substitution at position 84. J. Biol. Chem. 272 18550–18557. [DOI] [PubMed] [Google Scholar]
- Nichols, J.C., Vyas, N.K., Quiocho, F.A., and Matthews, K.S. 1993. Model of lactose repressor core based on alignment with sugar-binding proteins is concordant with genetic and chemical data. J. Biol. Chem. 268 17602–17612. [DOI] [PubMed] [Google Scholar]
- Ogata, R.T. and Gilbert, W. 1978. An amino-terminal fragment of lac repressor binds specifically to lac operator. Proc. Natl. Acad. Sci. 75 5851–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt, T., Files, J.G., and Weber, K. 1973. Lac repressor: Specific proteolytic destruction of the NH2-terminal region and loss of the deoxyribonucleic acid-binding activity. J. Biol. Chem. 248 110–121. [PubMed] [Google Scholar]
- Quiocho, F.A. and Vyas, N.K. 1984. Novel stereospecificity of the L-arabinose-binding protein. Nature 310 381–386. [DOI] [PubMed] [Google Scholar]
- Sams, C.F., Vyas, N.K., Quiocho, F.A., and Matthews, K.S. 1984. Predicted structure of the sugar-binding site of the lac repressor. Nature 310 429–430. [DOI] [PubMed] [Google Scholar]
- Schmitz, A., Schmeissner, U., Miller, J.H., and Lu, P. 1976. Mutations affecting the quaternary structure of the lac repressor. J. Biol. Chem. 251 3359–3366. [PubMed] [Google Scholar]
- Schumacher, M.A., Choi, K.Y., Zalkin, H., and Brennan, R.G. 1994. Crystal structure of LacI member, PurR, bound to DNA: Minor groove binding by α helices. Science 266 763–770. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Choi, K.Y., Lu, F., Zalkin, H., and Brennan, R.G. 1995. Mechanism of corepressor-mediated specific DNA binding by the purine repressor. Cell 83 147–155. [DOI] [PubMed] [Google Scholar]
- Slijper, M., Bonvin, A.M.J.J., Boelens, R., and Kaptein, R. 1996. Refined structure of lac repressor headpiece (1–56) determined by relaxation matrix calculations from 2D and 3D NOE data: Change of tertiary structure upon binding to the lac operator. J. Mol. Biol. 259 761–773. [DOI] [PubMed] [Google Scholar]
- Spott, S., Dong, F., Kisters-Woike, B., and Müller-Hill, B. 2000. Dimerisation mutants of lac repressor. II. A single amino acid substitution, D278L, changes the specificity of dimerisation. J. Mol. Biol. 296 673–684. [DOI] [PubMed] [Google Scholar]
- Suckow, J., Markiewicz, P., Kleina, L.G., Miller, J., Kisters-Woike, B., and Müller-Hill, B. 1996. Genetic studies of the lac repressor XV: 4000 Single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J. Mol. Biol. 261 509–522. [DOI] [PubMed] [Google Scholar]
- Swindells, M.B., Orengo, C.A., Jones, D.T., Hutchinson, E.G., and Thornton, J.M. 1998. Contemporary approaches to protein structure classification. Bioessays 20 884–891. [DOI] [PubMed] [Google Scholar]
- von Wilcken-Bergmann, B. and Müller-Hill, B. 1982. Sequence of galR gene indicates a common evolutionary origin of lac and gal repressor in Escherichia coli. Proc. Natl. Acad. Sci. 79 2427–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas, N.K., Vyas, M.N., and Quiocho, F.A. 1988. Sugar and signal-transducer binding sites of the Escherichia coli galactose chemoreceptor protein. Science 242 1290–1295. [DOI] [PubMed] [Google Scholar]
- Vyas, N.K., Vyas, M.N., and Quiocho, F.A. 1991. Comparison of the periplasmic receptors for L-arabinose, D-glucose/D-galactose, and D-ribose: Structural and functional similarity. J. Biol. Chem. 266 5226–5237. [PubMed] [Google Scholar]
- Weickert, M.J. and Adhya, S. 1992. A family of bacterial regulators homologous to Gal and Lac repressors. J. Biol. Chem. 267 15869–15874. [PubMed] [Google Scholar]
- Wycuff, D.R. 1999. "Examination of the C-terminal assembly motif in lactose repressor." Doctoral thesis. Rice University, Houston, Texas.