

Abstract
Complement is an important mediator of vascular injury following oxidative stress. We recently demonstrated that complement activation following endothelial oxidative stress is mediated by mannose-binding lectin (MBL) and activation of the lectin complement pathway. Here, we investigated whether nine plant lectins which have a binding profile similar to that of MBL competitively inhibit MBL deposition and subsequent complement activation following human umbilical vein endothelial cell (HUVEC) oxidative stress. HUVEC oxidative stress (1% O2, 24 hr) significantly increased Ulex europaeus agglutinin II (UEA-II) binding by 72 ± 9% compared to normoxic cells. UEA-II inhibited MBL binding to HUVEC in a concentration-dependent manner following oxidative stress. Further, MBL inhibited UEA-II binding to HUVEC in a concentration-dependent manner following oxidative stress, suggesting a common ligand. UEA-II (≤ 100 μmol/L) did not attenuate the hemolytic activity, nor did it inhibit C3a des Arg formation from alternative or classical complement pathway-specific hemolytic assays. C3 deposition (measured by ELISA) following HUVEC oxidative stress was inhibited by UEA-II in a concentration-dependent manner (IC50 = 10 pmol/L). UEA-II inhibited C3 and MBL co-localization (confocal microscopy) in a concentration-dependent manner on HUVEC following oxidative stress (IC50 ≈ 1 pmol/L). Finally, UEA-II significantly inhibited complement-dependent neutrophil chemotaxis, but failed to inhibit fMLP-mediated chemotaxis, following endothelial oxidative stress. These data demonstrate that UEA-II is a novel, potent inhibitor of human MBL deposition and complement activation following human endothelial oxidative stress.
Keywords: Hypoxia, reoxygenation, mannose-binding lectin, neutrophils, chemotaxis
Endothelial cells are important in the regulation of coagulation, vascular permeability, vasomotor tone and inflammation. Oxidative stress may result in complement activation and endothelial dysfunction. Endothelial dysfunction and complement activation are thought to be involved in human pathology including myocardial infarction (Weisman et al. 1990; Tsao et al. 1990), lung injury (Bless et al. 1999), sepsis (Czermak et al. 1999), and gut ischemia (Siegfried et al. 1992). Indeed, inhibition of complement attenuates tissue injury in patients undergoing cardiopulmonary bypass (Fitch et al. 1999) and in several experimental models of human disease (Weisman et al. 1990; Vakeva et al. 1998; Zhou et al. 2000). Complement is known to interact with the vascular endothelium and initiate a variety of pro-inflammatory signals, including leukocyte adhesion molecule expression, pro-inflammatory cytokine secretion, and loss of endothelium-dependent relaxation (Stahl et al. 1995; Kilgore et al. 1996; Collard et al. 1999; Buerke et al. 1998). Thus, the development of complement inhibitors may reduce endothelial dysfunction and tissue injury in a variety of clinical settings.
During the early stages of reperfusion of ischemic tissue, complement activation is known to occur initially at the endothelial cell (Weisman et al. 1990). We recently demonstrated that mannose-binding lectin (MBL) deposition and lectin complement pathway activation occurs on human endothelial cells following oxidative stress (Collard et al. 2000). MBL is a C-type lectin whose binding is calcium-dependent and has a high specificity to N-acetyl-D-glucosamine (GlcNAc), mannose or their oligomers (Thiel et al. 1997). Although the molecular mechanism by which oxidative stress increases MBL binding to endothelial cells is at present unclear, hypoxia alters endothelial protein synthesis and surface expression (Ogawa et al. 1991; Weinhouse et al. 1993; Dore-Duffy et al. 1999). Other lectins derived from plant sources have binding profiles similar to that of MBL. In the present study we investigated whether endothelial oxidative stress increases the binding of plant lectins displaying saccharide specificity similar to that of human MBL. We also investigated whether these lectins could be used to inhibit human MBL binding and lectin complement pathway activation following endothelial oxidative stress. We found that the plant lectin Ulex europaeus agglutinin II (UEA-II) significantly attenuates human MBL binding, lectin complement pathway activation, and the resulting complement-dependent neutrophil chemotaxis following endothelial oxidative stress.
Results
Lectin binding following endothelial oxidative stress
The plant lectins specific for GlcNAc, mannose or their oligomers used in this study are listed in Table 1. As shown in Figure 1 ▶, of the lectins screened, only UEA-II demonstrated a significant (P < 0.05) increase in binding to HUVEC following oxidative stress compared to normoxic cells. This observation is analogous to our recent finding that endothelial oxidative stress increases human MBL binding (Collard et al. 2000), as both UEA-II and MBL are calcium-dependent lectins with a high specificity for GlcNAc and its oligomers. The binding of UEA-II was unique, since the lectins LEA, STL, and WGA did not increase their binding to HUVEC following oxidative stress, yet have specificity for GlcNAc and its oligomers.
Table 1.
HRP-conjugated lectins used in this study
| Specificity | |||
| Lectin | GlcNAc | Oligomers | Abbreviation |
| Concanavalin A | − | − | Con A |
| Datura stramonium | − | − | DSA |
| Lycopersicin esculentum | + | + | LEA |
| Lens culinaris agglutinin | − | − | LCH |
| Ulex europaeus agglutinin-II | + | + | UEA-II |
| Griffonia simplicifonia | + | − | GSL |
| Solanum tuberosum | + | + | STL |
| Wheat germ agglutinin | ± | + | WGA |
| Succinylated wheat germ | + | − | SWGA |
Con A and LCH are specific for mannose (α-linked) residues and their binding is Ca++ and Mg++ dependent. Specificities summarized from http://www.vectorlabs.com and from the E-Y Laboratories, Inc. catalogue. E-Y Laboratories, Inc. was the source of all the lectins and HRP-conjugated lectins used in this study. GlcNAc = N-acetylglucosamine.
Fig. 1.

Lectin binding following HUVEC oxidative stress (ELISA). The effect of endothelial oxidative stress on lectin (10 μg/mL) binding was investigated by ELISA. Only UEA-II deposition following endothelial oxidative stress was significantly increased compared to normoxic cells. Data are presented as the percent change in lectin binding compared to normoxia with the data being normalized to the measured optical density (O.D.) of each respective lectin at baseline (normoxia). (n = 3; error bars = SEM; *P < 0.05 compared to normoxic cells; see Table 1 for lectin abbreviations).
UEA-II and MBL competition assays
We hypothesized that MBL and UEA-II may compete for a common ligand on HUVEC following oxidative stress (Fig. 2 ▶). The treatment of HUVEC with UEA-II significantly (P < 0.05) attenuated MBL binding in a concentration-dependent manner following oxidative stress (Fig. 2A ▶). Similarly, purified human MBL significantly (P < 0.05) attenuated UEA-II binding to HUVEC following endothelial oxidative stress (Fig. 2B ▶). These data suggest that MBL and UEA-II compete for a common HUVEC ligand following oxidative stress.
Fig. 2.
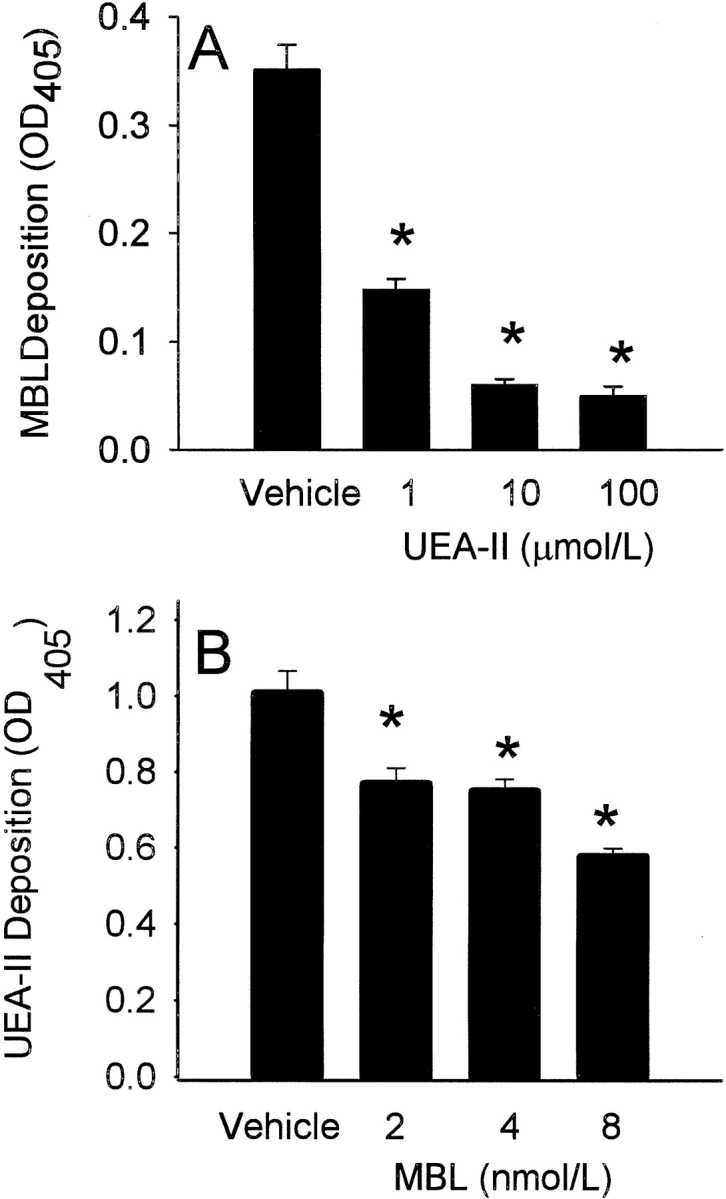
Competitive inhibition of MBL or UEA-II binding following HUVEC oxidative stress (ELISA). Competitive binding ELISA were performed in order to determine whether MBL and UEA-II compete for a common endothelial binding site following oxidative stress. (A) Treatment of HUVEC with UEA-II significantly (P < 0.05) attenuated MBL binding in a concentration-dependent manner following oxidative stress. (B) Addition of increasing amounts of purified human MBL significantly (P < 0.05) attenuated UEA-II binding to HUVEC following oxidative stress. (n = 3; error bars = SEM; *P < 0.05 compared to vehicle).
MBL and C3 deposition following endothelial oxidative stress
Endothelial oxidative stress increases MBL deposition, activates the lectin complement pathway, and deposits C3 (Collard et al. 2000). We investigated whether treatment of HUVEC with UEA-II decreases lectin complement pathway activation and C3 deposition following oxidative stress. As we have shown previously (Collard et al. 2000), GlcNAc (100 mmol/L) significantly inhibited C3 deposition on HUVEC exposed to oxidative stress (Fig. 3 ▶). C3 deposition following endothelial oxidative stress was significantly (P < 0.05) decreased in a concentration-dependent manner by the treatment of HUVEC with UEA-II (IC50 ≈10 pmol/L). Treatment of HUVEC (Fig. 4 ▶, B-E) with UEA-II (0.1 pmol/L–100 nmol/L) significantly decreased C3 and MBL deposition and colocalization in a concentration-dependent manner following oxidative stress compared to untreated cells. The results of these experiments collectively suggest that UEA-II potently attenuates endothelial MBL deposition and lectin complement pathway activation following oxidative stress.
Fig. 3.

UEA-II attenuates C3 deposition following endothelial oxidative stress (ELISA). C3 deposition following endothelial oxidative stress was measured by ELISA. C3 deposition following endothelial oxidative stress was significantly decreased in a concentration-dependent manner by treatment of HUVEC with UEA-II (IC50 ≈ 10 pmol/L). Data are normalized to the measured O.D. of untreated hypoxic/reoxygenated cells (O.D. at 450 nm = 0.77 ± 0.12) and expressed as percent inhibition. (n = 6, error bars = SEM, *P < 0.05 compared to vehicle).
Fig. 4.
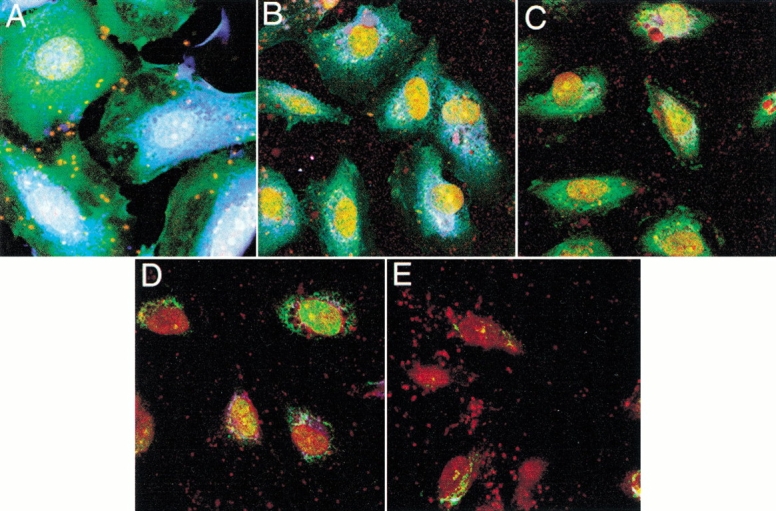
UEA-II attenuates MBL and C3 deposition following endothelial oxidative stress (confocal microscopy). Confocal microscopical demonstration of HUVEC MBL (blue) and C3 (green) deposition following oxidative stress. Treatment of HUVEC with UEA-II (B) 0.1 fmol/L; (C) 100 fmol/L; (D) 100 pmol/L; (E) 100 nmol/L UEA-II) decreased MBL and C3 deposition and co-localization (white) in a concentration-dependent manner compared to untreated cells (A) following oxidative stress. This figure is representative of three experiments. (Endothelial nuclei are indicated by red.)
Complement hemolytic and C3a assays
Complement hemolytic assays using sensitized chicken red blood cells (RBCs) were performed in order to demonstrate that UEA-II does not directly inhibit or activate (i.e., deplete) the classical complement pathway (Fig. 5 ▶). Treatment of human serum (HS) with UEA-II (1 μmol/L) did not significantly alter serum hemolytic activity compared to untreated HS. Thus, UEA-II does not directly inhibit or activate (i.e., deplete) the classical complement pathway.
Fig. 5.

UEA-II does not alter serum hemolytic activity. Complement hemolytic assays using sensitized chicken red blood cells were performed in order to demonstrate that UEA-II does not directly inhibit or activate complement. Treatment of HS with UEA-II (1 μmol/L) did not significantly alter hemolytic activity compared to untreated HS. (n = 2, error bars = SEM.)
In order to further demonstrate that UEA-II does not alter classical or alternative pathway activation, C3a des Arg measurements were performed in classical and alternative pathway-specific hemolytic assays. Rabbit RBCs were incubated with human sera in the presence of Mg2+/EGTA to allow alternative pathway activation (Lennon et al. 1996). Similarly, sensitized chicken RBCs were prepared as described in Figure 5 ▶. The data in Table 2 show that UEA-II (1 μmol/L) did not attenuate the production of C3a des Arg from the alternative pathway or the classical pathway. These results demonstrate that UEA-II does not alter alternative or classical complement pathway activation.
Table 2.
C3a des Arg measurements for classical and alternative specific hemolytic assays
| C3a des Arg concentration (ng/ml) | ||
| Pathway | Without UEA-II | With UEA-II |
| Alternative pathway | 8120 ± 965 | 7110 ± 1022 |
| Classical pathway | 3830 ± 722 | 3540 ± 493 |
UEA-II concentration was 1 μmol/L (1:30 HS diluted with GVB, see Materials and Methods) in both assays. Each value represents the mean ± SEM for 10 experiments. There was no significant difference between the C3a des Arg concentration in treated vs. untreated samples. Samples were assayed in duplicate.
Neutrophil chemotaxis following oxidative stress
Complement activation on endothelial cells leads to the generation of potent chemoattractants including the anaphylatoxin C5a, IL-8, and monocyte chemoattractant protein (Kilgore et al. 1996; Saadi et al. 2000). We hypothesized that UEA-II would decrease complement-mediated neutrophil chemotaxis following endothelial oxidative stress. As is shown in Figure 6 ▶, reoxygenation of hypoxic HUVEC in human serum (HS) significantly (P < 0.05) increased neutrophil chemotaxis compared to normoxic cells bathed in HS. The treatment of HUVEC with UEA-II (100 nmol/L) significantly attenuated neutrophil migration following endothelial oxidative stress compared to vehicle-treated cells. The treatment of normoxic cells with UEA-II had no effect on fMLP-driven chemotaxis compared to untreated normoxic HUVEC (data not shown), which indicates that the effect of UEA-II on chemotaxis is not a non-specific effect on neutrophil function. These data demonstrate that UEA-II attenuates complement-mediated neutrophil chemotaxis following endothelial oxidative stress.
Fig. 6.

Neutrophil chemotaxis is attenuated following oxidative stress by treatment of HUVEC with UEA-II. Oxidative stress significantly increased neutrophil chemotaxis compared to normoxic cells. Treatment of HUVEC with UEA-II (100 nmol/L) significantly attenuated neutrophil chemotaxis following endothelial oxidative stress. Neutrophil chemotaxis to HUVEC following oxidative stress was measured by analysis of myeloperoxidase levels and transformed to neutrophil count with a standard curve (n = 3, error bars = SEM, *P < 0.05 compared to normoxia, P < 0.05 compared to vehicle-treated hypoxia/reoxygenation cells).
Discussion
In this study, the binding of the calcium-dependent, GlcNAc-specific lectin UEA-II to HUVEC was increased following oxidative stress. UEA-II was found to inhibit MBL binding and C3 deposition in a potent and concentration-dependent manner. The finding that only one of the nine lectins assayed increased binding following oxidative stress can be explained by a previous finding that while lectins increased their binding to a few endothelial glycoproteins exposed to hypoxia, binding to the majority of endothelial glycoproteins was unchanged (Weinhouse et al. 1993). In addition, of the three lectins specific for GlcNAc and its oligomers (Table 1), UEA-II is the only leguminous lectin derived from seeds, the other two being derived from either potato tubers or the locular fluid of the tomato (Cummings 1999). The greater magnitude of the effect of UEA-II on MBL binding compared to that of MBL on UEA-II binding could be due to the greater molar ratios of UEA-II that were used, a greater affinity of UEA-II for the ligand(s), or both. We have thus obtained strong evidence that the plant lectin UEA-II is a potent lectin complement pathway inhibitor whose site of action is at the level of the endothelial cell, and whose molecular mechanism of action may be due to HUVEC expression of a common ligand for MBL and UEA-II following oxidative stress.
UEA-II is one of at least two lectins derived from the seeds of the Furze gorse plant (Matsumoto and Osawa 1969). UEA-II is a glycoprotein consisting of four 24,000 dalton monomer subunits that require Ca2+ for binding to its ligands via the carbohydrate recognition domain (Konami et al. 1992). Although the primary structures of UEA-I and UEA-II exhibit a high degree of homology (Konami et al. 1991), the two lectins have different carbohydrate specificity. UEA-I is specific for L-fucose, whereas UEA-II is specific for di-N-acetylchitobiose, an oligomer of GlcNAc (Matsumoto and Osawa 1969). Further, UEA-I is known to bind to endothelial cells under normal conditions, and since MBL binds to endothelial cells only following oxidative stress, UEA-I was not investigated in the present study. For these reasons, we chose to screen UEA-II and other lectins that were specific for GlcNAc, oligomers of GlcNAc, or mannose. Our intention was to find a plant lectin(s) that would bind to endothelial cells following oxidative stress like human MBL (e.g., increased binding following injury) and potentially inhibit lectin complement pathway activation. The results of the present study extend our previous finding that MBL is responsible for complement activation on endothelial cells following oxidative stress (Collard et al. 2000). We previously demonstrated the generation of functionally inhibitory antibodies against human MBL that inhibited complement activation on human endothelial cells following oxidative stress. In the present study, we identified UEA-II as a potent inhibitor of the lectin complement pathway following endothelial oxidative stress. The molecular mechanism of UEA-II-induced inhibition of the lectin complement pathway appears to be through competition for a common binding site on the endothelial cell and not at the level of MBL inhibition. MBL ligand inhibition may have a therapeutic advantage over complete inhibition of MBL, in that (a) one could specifically target the ligand responsible for MBL binding and not attenuate the additional functions associated with MBL (e.g., opsonization), and (b) site-directed complement inhibition would be localized to the area experiencing oxidative stress, and thus the amount of inhibitor utilized would be proportional of the size of the vascular bed.
Since the lectin complement pathway is activated by oxidative stress, and complement activation on endothelium leads to the formation of C5a as well as IL-8 and monocyte chemoattractant protein-1 secretion (Kilgore et al. 1996), we hypothesized that endothelial oxidative stress would lead to an increase in neutrophil chemotaxis that would be inhibited by UEA-II treatment. UEA-II treatment of HUVEC exposed to hypoxia/reoxygenation and HS significantly decreased neutrophil chemotaxis, suggesting a possible anti-inflammatory action of UEA-II. The anti-inflammatory effect of another GlcNAc-specific plant lectin was recently demonstrated in rat models of paw edema and peritonitis (Alencar et al. 1999). Further, plant lectin binding to endothelial cells has been shown to induce expression of CD59, an inhibitor of C5b-9 (e.g., pro-inflammatory molecule) formation (Dalmasso et al. 2000). Preliminary studies in our laboratory have demonstrated that UEA-II does not bind to rat endothelial cells following gastrointestinal or myocardial ischemia and reperfusion (data not shown). Like most protein-based complement inhibitors, plant lectins may also be species-specific inhibitors. We are currently screening plant lectins in vitro for their binding profiles on rat endothelial cells following oxidative stress. Thus, plant lectins may represent a novel class of complement inhibitors or regulators.
In summary, human endothelial oxidative stress increases UEA-II and MBL binding. UEA-II competes with human MBL for a common binding site on human endothelial cells. Treatment of endothelial cells with UEA-II decreases MBL deposition and complement activation in a concentration-dependent manner following oxidative stress. Finally, UEA-II treatment decreases complement-dependent neutrophil chemotaxis following endothelial oxidative stress. Together these data suggest that lectin therapy may represent a novel strategy for inhibiting complement-mediated injury in humans.
Materials and methods
Cell Culture
Human umbilical vein endothelial cells (HUVEC) were obtained as previously described (Collard et al. 1997). Endothelial cell purity was confirmed by a phase-microscopic "cobblestone appearance," uptake of fluorescent acetylated low-density lipoproteins, and the presence of von Willebrand factor. All experiments were conducted on cells in passages 1–3.
Lectin enzyme-linked immunosorbent assay (ELISA)
HUVEC were grown to confluence on 0.1% gelatinized 96-well plastic plates (Corning Costar, Cambridge, MA). The plates were then exposed to 0 (normoxia) or 24 hr of hypoxia (1% O2, 5% CO2, balance N2) at 37°C using a humidified sealed chamber (Coy Laboratory Products Inc., Grass Lake, MI) as described previously (Collard et al. 1997, 1999). The medium was then removed and the cells reoxygenated (21% O2, 5% CO2) for 3 hr at 37°C in 100 μL of Hanks Balanced Salt Solution (Sigma Chemical Co., St. Louis, MO) supplemented with 40 mmol/L Ca2+, Mg2+ and one of the HRP-conjugated (10 μg/mL) lectins (E-Y Laboratories, San Mateo, CA) listed in Table 1. The cells were then washed (NaCl 143 mmol/L, HEPES 10 mmol/L, CaCl2 40 mmol/L and MgCl2 40mmol/L) developed with ABTS [2,2'-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid)], and read at 405 nm on an automated plate reader (Molecular Devices, Sunnyvale, CA). This experiment was performed three times using five wells per experimental group (n = 3). Background optical density consisted of wells containing no cells and was subtracted from all groups.
In a separate experiment, the cell medium was removed following the specified period of normoxia or hypoxia and the cells were reoxygenated for 3 hr in the presence of 100 μL of one of the following: 1) 30% human serum (HS) diluted in calcium-supplemented gelatin veronal buffer (GVB); 2) 30% HS containing pure UEA-II (1,10, or 100 μmol/L, E-Y Laboratories); or 3) horseradish peroxidase (HRP)-conjugated UEA-II (100 μmol/L) in GVB containing purified human MBL (2, 4 or 8 nmol/L). MBL was purified as described (Tan et al. 1996; Collard et al. 2000). Cells reoxygenated in GVB (i.e., no sera) were washed and developed as described as above. Cells reoxygenated in HS were washed and lightly fixed with 1% paraformaldehyde (Sigma Chemical Co.) for 15 min. These cells were washed again and incubated at 4°C for 1 hr with 50 μL of peroxidase-conjugated rabbit anti-human MBL pAb (R2.2). The cells were then washed and developed as described above. Background optical density, subtracted from all groups, consisted of wells to which no HRP-conjugated UEA-II was added for experiments containing GVB only, or only the anti-human MBL antibody (i.e., no HS) for cells reoxygenated in HS. This experiment was performed three times using six wells per experimental group (n = 3).
C3 ELISA
HUVEC were grown to confluence on 96-well plastic plates and then subjected to 0 or 24 hr of hypoxia. Following the specified period of normoxia or hypoxia, the medium was aspirated and the cells reoxygenated (3 hr) in the presence of 100 μL of one of the following: 1) 30% HS diluted in GVB; 2) 30% HS with 0.1 pmol/L–100 nmol/L UEA-II; or 3) 30% HS with 100 mmol/L GlcNAc. The cells were then washed and fixed with 1% paraformaldehyde for 15 min. After washing, the cells were incubated for 1 hr at room temperature with 50 μL of HRP-conjugated goat anti-human C3 pAb (Cappell Laboratories, West Chester, PA) diluted 1 : 2,000 in 3% bovine serum albumin. The cells were washed and developed as described above. Background optical density was subtracted from all groups and consisted of cells to which HS was not added. This experiment was performed six times using six wells per experimental group (n = 6).
Complement hemolytic and C3a assays
HS was incubated with 0 or 1 μmol/L of UEA-II for 30 min at 37°C. The HS was then diluted serially 1 : 2 (v : v) in GVB. Classical complement pathway hemolytic assays using sensitized chicken RBCs were performed as previously described (Vakeva et al. 1998). This experiment was performed in triplicate twice (n = 2).
Classical pathway-specific C3a des Arg generation was performed using sensitized chicken RBCs as described above. HS was diluted 1 : 30 with GVB and then treated with UEA-II (1 μmol/L) or vehicle (phosphate-buffered saline (PBS)). This concentration of HS induces about 50% lysis of sensitized cells (Fig. 5 ▶).
Alternative pathway-specific C3a des Arg generation was performed as described using rabbit RBCs (Lennon et al. 1996). HS was diluted to 1 : 30, as this concentration induced approximately 50% lysis of rabbit RBCs in pilot studies, then treated with UEA-II (1 μmol/L) or vehicle (PBS). C3a des Arg was measured with a commercially available kit (OptiEIA, BD Pharmingen, San Diego, CA).
Immunofluorescent confocal microscopy
HUVEC grown on Labtek (NUNC) tissue culture slides were subjected to 0 or 24 hr of hypoxia and then reoxygenated for 3 hr in the presence of GVB, 30% HS or 30% HS treated with UEA-II (0.1 fmol/L-100 nmol/L). The cells were then washed twice, fixed with 1% paraformaldehyde, and washed again. The cells were then incubated overnight (4°C) with a biotinylated mouse anti-human MBL mAb (clone 1C10) (Collard et al. 2000) and FITC-conjugated F(ab)2 goat anti-human C3 pAb (ICN, Aurora, OH). The cells were washed and incubated (2 hr at room temperature) with streptavidin-conjugated CY5 (1 μg/mL, Jackson Immunoresearch, West Grove, PA). The cells were also incubated with propidium iodide (20 μg/mL) for 10 min to stain the nuclei. After being washed, the slides were coated with anti-fade mounting medium (Molecular Probes, Eugene, OR), covered, and analyzed with a Leica confocal laser-scanning microscope as previously described (Collard et al. 2000). All analyses were conducted at the same pinhole, voltage and laser settings.
Neutrophil chemotaxis assay
HUVEC were grown to confluence on 24-well plates and then subjected to 0 or 24 hr of hypoxia. Following the specified period of normoxia or hypoxia, the medium was aspirated and the cells were reoxygenated (3 hr) in the presence of 30% HS or 30% HS treated with UEA-II (100 nmol/L). During the reoxygenation period, human neutrophils were harvested and isolated as previously described (Henson and Oades 1975). Five-micron transwell inserts (Corning Costar, Cambridge MA) were then placed in each well of the reoxygenated HUVEC. Human neutrophils (2 × 106 cells/well) were added to each transwell and incubated for 90 min at 37°C. The supernatant covering the HUVEC was removed and centrifuged at 150 × g for 10 minutes. The resulting pellet was resuspended in 1 mL of Hanks Balanced Salt Solution, solubilized with 50 μL of 10% Triton X -100 and acidified with 100 μL of citrate buffer (1 mol/L pH 6.5). The myeloperoxidase content of the wells was then assayed as previously described (Parkos et al. 1992). The absolute neutrophil count was determined by using a standard curve. This experiment was performed three times with three wells per experimental group (n = 3).
Statistical analysis
Data analyses were performed using Sigma Stat (Jandel Scientific, San Rafael, CA). Neutrophil counts and lectin deposition on normoxic versus hypoxic HUVEC were analyzed by two-way analysis of variance (ANOVA). C3 deposition was analyzed by one-way ANOVA. All pairwise multiple comparisons were made using the Student-Newman-Keuls test. The C3a des Arg concentrations were analyzed by Student's t-test. All data are expressed as means ± SEM. Probability values < 0.05 were considered significant.
Acknowledgments
Sources of support for these studies included NIH grants GM-07592 (RL), HL-03854 (CDC), HL-52886 (GLS), and HL-56086 (GLS). Dr. Stahl is an Established Investigator of the American Heart Association. We thank Margaret Morrissey and Meera Grover.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
fMLP, formyl methionine leucine phenylalanine
GlcNAc, N-acetylglucosamine
GVB, gelatin veronal buffer
HRP, horseradish peroxidase
HS, human serum
HUVEC, human umbilical vein endothelial cells
mAb, monoclonal antibody
MBL, mannose-binding lectin
pAb, polyclonal antibody
UEA-II, Ulex europaeus agglutinin II.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.26401.
References
- Alencar, N.M., Teixeira, E.H., Assreuy, A.M., Cavada, B.S., Flores, C.A., and Ribeiro, R.A. 1999. Leguminous lectins as tools for studying the role of sugar residues in leukocyte recruitment. Mediators Inflamm 8 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bless, N.M., Warner, R.L., Padgaonkar, V.A., Lentsch, A.B., Czermak, B.J., Schmal, H., Friedl, H.P., and Ward, P.A. 1999. Roles for C-X-C chemokines and C5a in lung injury after hindlimb ischemia-reperfusion. Am J Physiol 276 L57–L63. [DOI] [PubMed] [Google Scholar]
- Buerke, M., Prüfer, D., Dahm, M., Oelert, H., Meyer, J., and Darius, H. 1998. Blocking of classical complement pathway inhibits endothelial adhesion molecule expression and preserves ischemic myocardium from reperfusion injury. Journal of Pharmacology and Experimental Therapeutics 286 429–438. [PubMed] [Google Scholar]
- Collard, C.D., Agah, A., Reenstra, W.R., Buras, J., and Stahl, G.L. 1999. Endothelial nuclear factor-kB translocation and vascular cell adhesion molecule-1 induction by complement: Inhibition with anti-human C5 therapy or cGMP analogues. Arterioscler Thromb Vasc Biol 19 2623–2629. [DOI] [PubMed] [Google Scholar]
- Collard, C.D., Vakeva, A., Bukusoglu, C., Zund, G., Sperati, C.J., Colgan, S.P., and Stahl, G.L. 1997. Reoxygenation of hypoxic human umbilical vein endothelial cells (HUVECs) activates the classic complement pathway. Circulation 96 326–333. [DOI] [PubMed] [Google Scholar]
- Collard, C.D., Vakeva, A., Morrissey, M.A., Agah, A., Rollins, S.A., Reenstra, W.R., Buras, J.A., Meri, S., and Stahl, G.L. 2000. Complement activation following oxidative stress: Role of the lectin complement pathway. Am J Pathol 156 1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings, R.D. 1999. Plant Lectins. In Essentials of Glycobiology, (eds. A. Varki et al.) pp. 455–469. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Czermak, B.J., Sarma, V., Pierson, C.L., Warner, R.L., Huber-Lang, M., Bless, N.M., Schmal, H., Friedl, H.P., and Ward, P.A. 1999. Protective effects of C5a blockade in sepsis. Nat Med 5 788–792. [DOI] [PubMed] [Google Scholar]
- Dalmasso, A.P., Benson, B.A., Johnson, J.S., Lancto, C., and Abrahamsen, M.S. 2000. Resistance against the membrane attack complex of complement induced in porcine endothelial cells with a galα(1–3)gal binding lectin: Up-regulation of CD59 expression. J Immunol 164 3764–3773. [DOI] [PubMed] [Google Scholar]
- Dore-Duffy, P., Balabanov, R., Beaumont, T., Hritz, M.A., Harik, S.I., and LaManna, J.C. 1999. Endothelial activation following prolonged hypobaric hypoxia. Microvasc Res 57 75–85. [DOI] [PubMed] [Google Scholar]
- Fitch, J.C.K., Rollins, S.A., Matis, L.A., Alford, B.L., Aranki, S., Collard, C.D., Dewar, M., Elefteriades, J., Hines, R., Kopf, G., et al. 1999. Pharmacology and biological efficacy of a recombinant, humanized, single chain antibody, C5 complement inhibitor in patients undergoing coronary artery bypass graft surgery utilizing cardiopulmonary bypass. Circulation 100 2499– 2509. [DOI] [PubMed] [Google Scholar]
- Henson, P.M. and Oades, Z.G. 1975. Stimulation of human neutrophils by soluble and insoluble immunoglobulin aggregates. Secretion of granule constituents and increased oxidation of glucose. J Clin Invest 56 1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore, K.S., Flory, C.M., Miller, B.F., Evans, V.M., and Warren, J.S. 1996. The membrane attack complex of complement induces interleukin-8 and monocyte chemoattractant protein-1 secretion from human umbilical vein endothelial cells. Am J Pathol 149 953–961. [PMC free article] [PubMed] [Google Scholar]
- Konami, Y., Yamamoto, K., and Osawa, T. 1991. The primary structures of two types of the Ulex europeus seed lectin. J Biochem (Tokyo) 109 650–658. [DOI] [PubMed] [Google Scholar]
- Konami, Y., Yamamoto, K., and Osawa, T. 1992. Purification and characterization of carbohydrate-binding peptides from Lotus tetragonolobus and Ulex europeus seed lectins using affinity chromatography. J Chromatogr 597 213–219. [DOI] [PubMed] [Google Scholar]
- Lennon, P.F., Collard, C.D., Morrissey, M.A., and Stahl, G.L. 1996. Complement-induced endothelial dysfunction in rabbits: mechanisms, recovery, and gender differences. Am J Physiol Heart Circ Physiol 270 H1924–H1932. [DOI] [PubMed] [Google Scholar]
- Matsumoto, I. and Osawa, T. 1969. Purification and characterization of an anti-H(O) phytohemagglutinin of Ulex europeus. Biochim Biophys Acta 194 180–189. [DOI] [PubMed] [Google Scholar]
- Ogawa, S., Clauss, M., Kuwabara, K., Shreeniwas, R., Butura, C., Koga, S., and Stern, D. 1991. Hypoxia induces endothelial cell synthesis of membrane- associated proteins. Proc Natl Acad Sci USA 88 9897–9901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkos, C.A., Colgan, S.P., Delp, C., Arnaout, M.A., and Madara, J.L. 1992. Neutrophil migration across a cultured epithelial monolayer elicits a biphasic resistance response representing sequential effects on transcellular and paracellular pathways. J Cell Biol 117 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadi, S., Holzknecht, R.A., Patte, C.P., and Platt, J.L. 2000. Endothelial cell activation by pore-forming structures: Pivotal role for interleukin-1alpha [In Process Citation]. Circulation 101 1867–1873. [DOI] [PubMed] [Google Scholar]
- Siegfried, M.R., Ma, X., and Lefer, A.M. 1992. Splanchnic vascular endothelial dysfunction in rat endotoxemia: Role of superoxide radicals. Eur J Pharmacol 212 171–176. [DOI] [PubMed] [Google Scholar]
- Stahl, G.L., Reenstra, W.R., and Frendl, G. 1995. Complement mediated loss of endothelium-dependent relaxation of porcine coronary arteries. Role of the terminal membrane attack complex. Circ Res 76 575–583. [DOI] [PubMed] [Google Scholar]
- Tan, S.M., Chung, M.C.M., Kon, O.L., Thiel, S., Lee, S.H., and Lu, J. 1996. Improvements on the purification of mannan-binding lectin and demonstration of its Ca2+-independent association with a C1s- like serine protease. Biochem J 319 329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel, S., Vorup-Jensen, T., Stover, C.M., Schwaeble, W., Laursen, S.B., Poulsen, K., Willis, A.C., Eggleton, P., Hansen, S., Holmskov, U., et al. 1997. A second serine protease associated with mannan-binding lectin that activates complement. Nature 386 506–510. [DOI] [PubMed] [Google Scholar]
- Tsao, P.S., Aoki, N., Lefer, D.J., Johnson III, G., and Lefer, A.M. 1990. Time course of endothelial dysfunction and myocardial injury during myocardial ischemia and reperfusion in the cat. Circulation 82 1402–1412. [DOI] [PubMed] [Google Scholar]
- Vakeva, A., Agah, A., Rollins, S.A., Matis, L.A., Li, L., and Stahl, G.L. 1998. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion. Role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 97 2259–2267. [DOI] [PubMed] [Google Scholar]
- Weinhouse, G.L., Belloni, P.N., and Farber, H.W. 1993. Effect of hypoxia on endothelial cell surface glycoprotein expression: Modulation of glycoprotein IIIa and other specific surface glycoproteins. Exp Cell Res 208 465–478. [DOI] [PubMed] [Google Scholar]
- Weisman, H.F., Bartow, T., Leppo, M.K., Marsh, H.C.J., Carson, G.R., Concino, M.F., Boyle, M.P., Roux, K.H., Weisfeldt, M.L., and Fearon, D.T. 1990. Soluble human complement receptor type 1: In vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 249 146–151. [DOI] [PubMed] [Google Scholar]
- Zhou, W., Farrar, C.A., Abe, K., Pratt, J.R., Marsh, J.E., Wang, Y., Stahl, G.L., and Sacks, S.H. 2000. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest 105 1363–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]