

Abstract
Mouse urine contains an abundance of major urinary proteins, lipocalins, whose roles include slow release of semiochemicals. These proteins are highly polymorphic, with small sequence differences between individual members. In this study, we purified to homogeneity four of these proteins from two strains of inbred mice and characterized them by mass spectrometry. This analysis has led to the discovery of another variant in this group of proteins. Three of the polymorphic variants that map to the surface have no effect on the binding of a fluorescent probe in the binding cavity, but the fourth, characterized by a Phe to Val substitution in the cavity, shows a substantially lower affinity and fluorescence yield for the probe. These results are interpreted in light of the known crystal structure of the protein and molecular modeling calculations, which rationalize the experimental findings. This work raises the possibility that the calyx-binding site can show specificity for different ligands, the implications of which on pheromone binding and chemical communication are discussed.
Keywords: Major urinary proteins, N-phenyl-naphthylamine, mass spectrometry, protein polymorphism
The urine of mice and other rodent species is unusual in that it manifests an obligate proteinuria. This proteinuria comprises a specific class of lipocalins, termed, in the mouse, major urinary proteins (MUPs), that are synthesized in the liver and subsequently pass through the glomerular filter to be released in the urine (Cavaggioni et al. 1999). These proteins have a typical lipocalin structure, comprising an eight stranded β-barrel that surrounds a central hydrophobic calyx (Bocskei et al. 1992; Flower 1996). The calyx of the MUPs binds semiochemicals (Bacchini et al. 1992; Robertson et al. 1993), and we have shown recently, both biochemically and by behavioral assays, that one outcome of this binding is to act as a slow-release mechanism, to allow the chemosensory information in urine to be extended in the time domain (Hurst et al. 1998; Beynon et al. 1999).
However, a simple role of ligand binding and slow release fails to account for the observation of the rather marked polymorphism that is a feature of the MUP family. To date, we have provided evidence for more than 50 different MUP variants (Robertson et al. 1996, 1997; Pes et al. 1999). For those variants for which the sequences are known, most of the amino acid substitutions are located on the surface of the molecule, but there are a limited number that alter the shape or environment of the central cavity. The role of these polymorphic variants is not known, and the predominantly surface location of the amino acid substitutions is not commensurate with their role as pheromone-binding proteins. Evidence is accumulating for receptors to MUP-type proteins, however (Krieger et al. 1999), and surface variations might be critical in MUP–receptor interactions. Certainly, MUPs in the absence of ligands elicit behavioral responses (Humphries et al. 1999).
The proven role of the proteins in ligand release suggests that the internal polymorphic variants also might have different binding characteristics for molecules that occupy the central calyx. As part of an analysis of the functional consequences of the MUP polymorphisms, we have discovered that these proteins are able to bind the fluorescent probe N-phenyl-naphthylamine (NPN), which is an effective optical probe of the MUP structure. NPN fluoresces weakly in an aqueous environment, but becomes strongly fluorescent in nonpolar or hydrophobic environments (Brito and Vaz 1986; Ocaktan et al. 1997) such as the calyx of the MUPs. In this report, we describe the binding of NPN to several MUP variants in which the sequence substitutions are located either on the protein surface or in the central cavity.
Results and discussion
Individual MUPs were purified from the urine of two inbred strains of mice, BALB/C and C57BL/6J by high-resolution ion exchange chromatography (results not shown). The nomenclature of the urinary MUPs was according to that used in Robertson et al. (1996). The purity of each of these proteins, and the lack of any postexcretory degradation, was confirmed by SDS-PAGE and electrospray ionization mass spectrometry. We searched for a suitable optical probe to monitor the cavity of MUPs. A series of charged naphthalene derivatives, including 1-anilinonaphthalene-8-sulphonate (1,8-ANS), 2-anilinonaphthalene-6-sulphonate acid (2,6-ANS), (2-(ρ-toluidinyl) naphthalene-6-sulfonic acid (2,6-TNS), and other probe (Nile Red) showed no change in fluorescence when added to MUPs, but one, NPN, gave a remarkable enhancement of fluorescence. This molecule has been used as a neutral probe of membrane environments (Brito and Vaz 1986; Ocaktan et al. 1997), and it fluoresces strongly when transferred from a polar to a nonpolar environment. The fluorescence enhancement was ∼50-fold and produced a blue shift to give an emission maximum at 395 nm. The fluorescence yield differed between individual MUPs, however, and in one variant in particular the fluorescence enhancement was substantially lower than the others (Fig. 1 ▶).
Fig. 1.

Fluorescence of N-phenyl-1-naphthylamine bound to purified polymorphic variants of major urinary proteins. MUPs, purified from BALB/C (uMUP-I and uMUP-VIII) or C57BL/6J (uMUP-IX and uMUP-X) were incubated at 50 nM final concentration with 50 nM NPN (structure inset) for 15 min before scanning the emission spectrum (excitation 377 nm). The control scan was of NPN dissolved in buffer at the same concentration.
The specificity of NPN binding was confirmed by displacement studies, in which NPN binding was monitored by both the increase in fluorescence and the displacement of a natural ligand, 2-sec-butyl-4,5-dihydrothiazole, by gas chromatography/mass spectrometry (Fig. 2 ▶). After incubation of a desalted mixed MUP preparation with NPN, the thiazole was no longer protein bound and could be separated by gel filtration. Commensurately, the enhancement of NPN fluorescence confirmed the binding of the probe to the protein. Thus, NPN binds selectively to the central calyx of the protein and displaces the natural ligand. Scatchard analysis of the NPN titration data for all four MUP variants in this study revealed a stoichiometry of near unity: 0.96 ± 0.018 (mean ±SD, n = 4, data not shown). NPN therefore is a specific probe for the internal environment of the MUP cavity.
Fig. 2.

Displacement of natural ligands on binding of NPN to MUPs. Desalted urinary MUPs (100 μM) were incubated with increasing concentrations of NPN. After incubation for 30 min, the MUP preparation was passed through a Sephadex G25 spun column. The protein fraction then was analyzed by GC/MS for bound 2-sec-butyl-4,5-dihydrothiazole, and an additional sample was analyzed for bound NPN by fluorescence. The 2-sec-butyl-4,5-dihydrothiazole abundance (filled squares) is expressed as a percentage of the amount protein-bound in the absence of NPN. Fluorescence (open circles) is expressed as arbitrary units.
We used the probe to explore the environment of a series of MUP polymorphic variants, purified from the urine of different inbred laboratory strains. Many of these variants have very similar properties and are not readily resolved by chromatography to single species (Robertson et al. 1996; Pes et al. 1999). However, we were able to purify to homogeneity four MUPs (from urine of BALB/C or C57BL/6J mice) by Resource Q fast protein liquid chromotography. One of these, uMUP-X from C57BL/6J mice, has no known cDNA or amino acid sequence, and we therefore characterized that protein by endopeptidase Lys-C fragmentation and MALDI-TOF mapping of the protein, including the other purified MUPs for comparison (Fig. 3 ▶). Endopeptidase Lys-C generates up to 11 fragments, only two of which (L3 and L6) are too small be seen by MALDI-TOF. If these two small fragments are assumed to be present in every MUP so far characterized, then the mass of the intact proteins measured by ESI-MS is exactly the same as that obtained from the sum of the masses of the constituent peptides. There therefore is no missing mass in these analyses. The previously uncharacterized uMUP-X has one peptide (L5) that differs from the "standard" map obtained for uMUP-I (Fig. 4 ▶). The mass of L5 is 2048.0 daltons, compared with 2095.9 daltons in the other proteins. This mass shift, of 47.9 daltons, is explicable by either a single Phe → Val amino acid change or a Tyr → Asp change. However, there is no tyrosyl residue in this peptide, whereas a valyl residue at position 56 has been observed previously in uMUP-VII from BALB/C mice. It therefore is highly likely that this smaller peptide is L5(F56V). uMUP-X and uMUP-VII both contain Val at position 56, but only uMUP-X can be purified to homogeneity.
Fig. 3.

MALDI-TOF mass spectrometry of Lys-C digests of purified MUP variants. MUPs, purified from BALB/C (uMUP-I and uMUP-VIII) or C57BL/6J (uMUP-IX and uMUP-X), were reduced and denatured and subjected to endopeptidase Lys-C digestion before MALDI-TOF mass spectrometry. The spectral peaks labeled with masses correspond to assigned MUP fragments (see Fig. 5 ▶). (Peaks labeled with a filled circle are methionine-oxidized variants of the adjacent, unoxidized peptide (Δm = 15.99 daltons), which are a coincidental confirmation of methionine-containing peptides.
Fig. 4.

Peptide mapping of MUP variants. MUPs, purified from BALB/C (uMUP-I and uMUP-VIII) or C57BL/6J (uMUP-IX and uMUP-IX), were reduced and denatured and subjected to endopeptidase Lys-C digestion before MALDI-TOF mass spectrometry. The amino acids at variant positions 50, 56, and 140 are highlighted. The peptide map at the top of the diagram is the theoretical map predicted from the cDNA-inferred sequence of uMUP-I; the other four maps are observed peptide masses. Where peptides differ from the "standard" uMUP-I map, they are appropriately labeled on the diagram (L4A, L4B, L5′, L10 + 11). The sum of the peptide masses (adjusted for water of hydrolysis) is compared with the equivalent monoisotopic mass (M + H) of the protein sequence that is derived from the predicted sequence.
Thus, the four proteins differ in just three positions in the entire sequence alignment: uMUP-I:N50F56K140, uMUP-VIII: N50F56E140, uMUP-IX: K50F56E140, uMUP-X: N50V56K140. Two of these positions (50 and 140) are located on the surface of the MUP (Fig. 5 ▶). Residue 140 is located on the solvent exposed face of the C-terminal α helix. Residue 50 is located at a sharp turn on the opposite face of the protein to the helical segment. In both positions, the amino acids are conservative, polar/polar changes, E/Q and K/N, respectively, and would not be expected to cause major structural shifts. In contrast, the remaining variant position 56 is located within the central calyx, and the Phe to Val substitution might be anticipated to elicit some difference in the ability to bind ligands. Accordingly, we used NPN as a probe to measure the ability of the different variants to bind ligands (Fig. 6 ▶). Titration of the four proteins with NPN showed that all three of the surface variants bound NPN in the same fashion, and the three binding curves were virtually superimposable. The Kd (fitted value ±SE) values were uMUP-I: 60.2 ± 2.4 nM, uMUP-VIII: 60.9 ± 2.5 nM, uMUP-IX: 56.2 ± 3.3 nM. In contrast, the Val 56 variant gave a much lower fluorescence yield and showed much weaker binding (uMUP-X: 122.3 ± 12.5nM). NPN binds some 20-fold tighter than the natural ligand 2-sec-butyl-4,5-dihydrothiazole (Zidek et al. 1999), and the Kdapp values determined here will not vary significantly from the true values.
Fig. 5.

Locations of polymorphic variants of uMUPs. The structure is drawn from the coordinates published by Bocskei et al. (1992). The variant positions of the four polymorphic forms of uMUPs are highlighted on the structure, as is the location of the natural ligand, 2-sec-butyl-4,5-dihydrothiazole (TZL); however, note that a recent NMR structure (Zidek et al. 1999) has provided some data for a different orientation of the ligand in the cavity.
Fig. 6.

NPN binding to MUP polymorphic variants. NPN (0–1500 nM) was titrated against 50-nM solutions of different MUPs purified from BALB/C (uMUP-I and uMUP-VIII) or C57BL/6J (uMUP-IX and uMUP-X). The binding isotherms were analyzed by nonlinear curve fitting as described in the experimental section. (circles) uMUP-X; (triangles) uMUP-I; (squares) uMUP-IX; (diamonds) uMUP-VIII. The bottom panel shows the same data normalized with the maximum fluorescence value set to 100%, to emphasize the difference in NPN binding ability of the variants.
The binding of NPN to various polymorphic variants was investigated further by molecular modeling calculations. A three-dimensional model of uMUP-X was constructed, based on the known crystal structure 1MUP.PDB, which has the highest amino acid sequence identity to uMUP-II (Fig. 7 ▶). Critically, the structure contains a phenylalanine residue at position 56 in the cavity interior. The model of uMUP-X (Val 56) possesses an altered cavity shape that is smaller (516 Å3) than the corresponding cavity in the crystal structure (526 Å3). The likely binding modes of NPN were explored by Monte Carlo molecular mechanics binding simulations. The lowest energy conformers, representing the likely binding conformations, are shown in Figure 8 ▶. Two distinctly different binding modes were observed. This is consistent with the fluorescence enhancement data, as the fluorescence yield is considerably higher in the Phe 56 variant (uMUP-II), in keeping with the rather more pronounced ring stacking interaction between this residue and NPN, which is observed in the modeled binding mode. Furthermore, the ICM software correctly predicts a tighter binding in the uMUP-II complex compared with the uMUP-X complex, consistent with the experimentally determined Kd values. The volume of NPN is 230 Å3, and we considered the possibility of a water molecule being bound within the cavity in addition to NPN, particularly in uMUP-X (Val 56) in which water quenching might diminish the fluorescence yield. In neither uMUP-II (Phe 56) nor uMUP-X (Val 56) is there a location where a 30-Å3 sphere (the approximate volume of a water molecule) can be comfortably accommodated. Moreover, the cavity is extremely hydrophobic in both proteins (>90% of the internal cavity surface is nonpolar). However, modeling studies cannot predict with certainty additional firmly bound water molecules, and their outcome should always be interpreted with caution.
Fig. 7.

Alignment of four polymorphic uMUP sequences. The four uMUP sequences studied in this investigation are aligned, to indicate cavity-binding residues (shaded), secondary structure (illustrated under the alignment), and the positions of the three polymorphic positions relating the four variants (asterisks). Of these, only position 56 is a cavity-lining residue.
Fig. 8.

Comparison of modeled NPN binding to two cavity polymorphic uMUP variants. The binding of NPN to uMUP-II and uMUP-X was modeled by Monte Carlo simulation using the ICM program. Both simulations converged to the distinct solutions that are shown in the figure.
Thus, cavity mutants of the MUPs show a marked difference in the ability to bind nonphysiological ligands, as measured by this optical probe. It remains to be seen whether this difference in binding ability is also manifest as a different affinity for, or specifically, rate of release of, natural semiochemical ligands. Although the surface mutations were without effect on the binding properties of the cavity, there remains the possibility that these have a functional significance in some other role, in which the proteins communicate information in their own right (Mucignat-Caretta et al. 1995; Humphries et al. 1999; Brennan et al. 1999). Finally, NPN binds so tightly and gives such a strong fluorescence yield that it might find value in the visualization or quantitation of MUPs in natural or seminatural experimental environments.
Materials and methods
Urine was collected from unanesthetized BALB/C or C57BL/6J male mice by gentle bladder massage and stored at −20°C until use. The urine subsequently was defrosted, and the protein fraction was separated from the low molecular weight components of urine by desalting over G25 "spun columns" equilibrated in 50 mM MES buffer at pH 5.0. The protein fraction was filtered through a 0.22-μm filter and loaded onto a MonoQ anion exchange column, previously equilibrated in 50 mM MES buffer at pH 5.0. After washing with three column volumes of the same buffer, the column then was developed by a linear gradient of between 0 and 200 mM NaCl, dissolved in the same buffer, over a total volume of 154 mL, and fractions of 2.0 mL were collected. Typically, 10 mg of protein, derived from ∼1.0 mL of urine, was applied to the column. Individual MUP fractions were pooled, and the purity and identity were confirmed by electrospray ionization mass spectrometry.
The structures of the individual MUPs were investigated further by peptide mapping of endopeptidase Lys-C peptides on MALDI-TOF mass spectrometry. Individual MonoQ fractions containing MUPs were precipitated with 5 M HCl at 4°C. The pellet was redissolved in 50 μL of 25 mM Tris buffer, 1 mM EDTA, 2 mM β-mercaptoethanol at pH 8.5, and Lys-C (5 μL of 0.5 μg/μL stock) was added, before overnight incubation at 37°C. At the end of this period, 10 μL formic acid was added. Peptides were analyzed by a PE Biosystems Voyager Elite laser desorption ionization time-of-flight mass spectrometer with an ion mirror (3-m path length) and delayed extraction function, equipped with a nitrogen laser (337 nm). In all experiments, mass spectrometry was performed in reflectron mode with positive ion detection. Instrument parameters were as follows: accelerating voltage 20 kV, grid voltage 73.5%, guide wire voltage 0.05%, and pulse delay time 175 nsec. External mass calibration was performed with a mixture of des-Arg bradykinin, neurotensin, adrenocorticotropic hormone, and insulin β-chain (50 nM each in 50% (v/v) acetonitrile/0.1% (v/v) triflouroacetic acid). To 1 μL of digested protein or peptide calibrants, 1 μL of α-cyano-4-hydroxy cinnamic acid (saturated in 50% (v/v) acetonitrile/0.1% (v/v) triflouroacetic acid) was added, and 1 μL was spotted onto the target. Signal averaged mass spectra for 80–256 laser shots were collected, and data were analyzed using the PE Biosystems GRAMS/386 software.
Binding studies were performed on individual MUPs with the fluorescent probe NPN. A stock solution of NPN (150 μM) was prepared by dissolving in dimethylformamide, the concentration of which was determined from the absorbance at 337 nm (E337 = 8100 M−1 cm−1). A solution of MUPs (50 nM) in 50 mM HEPES at pH 7.5 was titrated by increasing additions of NPN and the fluorescence (excitation 337 nm; emission 395 nm) was measured at 20°C using a Perkin-Elmer LS50B spectrofluorimeter, 15 min after addition of NPN. All readings were signal-averaged for 15 sec. No correction for inner filter effects was necessary. Fluorescence intensity was plotted versus increasing NPN concentration, and nonlinear least-squares analysis (FigP version 2.98, Biosoft) was applied to calculate the dissociation constants for different MUP isoforms by using the model described in Epps et al. (1995):
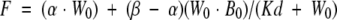 |
where F is the measured fluorescence, α and β are the proportionality constants, W0 is the total ligand concentration, B0 is the total protein concentration, and Kd is the dissociation constant. The data are presented as theoretical best-fit lines superimposed on experimental data.
The displacement of natural ligands by NPN was assessed by gas chromatography/mass spectrometry (GC/MS). MUPs (100 μM) were incubated with increasing concentrations of NPN from stock solutions in ethanol. After a 30-min incubation, the mixture was desalted using Sephadex G25 spun columns, and 200 μL of the desalted samples was taken for headspace sampling and GC/MS analysis. Headspace sampling was achieved using a Hewlett-Packard HP 7694E headspace analyzer. Samples were sealed in 10-mL crimp-top vials, which then were heated for 20 min at 100°C, after which a 3-mL headspace gas sample was taken and transferred to the GC/MS via a heated transfer line. Both sample loop and transfer line were maintained at a temperature of 120°C. Carrier gas and vial pressurization gas (He in both cases) were maintained at pressures recommended by the manufacturer. The headspace sample was introduced into the split inlet port of a Hewlett-Packard 5890 GC, fitted with a Hewlett Packard 5791A mass selective detector. The injector was maintained at a temperature of 250°C, and a split ratio of 100:1 was used to achieve a final flow rate of 1 mL/min through the column. In all analyses, the GC column used was a ZB-wax column (Phenomenex), 30 m in length, 0.25 mm i.d., and 0.25 μm film thickness. The column was maintained at 70°C for 2 min before being increased to 200°C at 10°C/min. The column was maintained at this temperature for an additional 2 min. The Hewlett Packard 5791A mass selective detector was used in selected ion monitoring mode for m/z 60 (2-sec-butyl-4,5-dihydrothiazole). Machine control, data acquisition, and analysis were all computer controlled using Hewlett-Packard ChemStation software. The 2-sec-butyl-4,5-dihydrothiazole was quantitated by peak area and expressed relative to the sample not treated with NPN. An additional 5 μL of each desalted sample was diluted to 3 mL in a fluorimeter cuvette, and the NPN fluorescence was measured as described above.
A tertiary structure model of uMUP-X was built using the MODELLER package (Sali and Blundell 1993) using the crystal structure (databank code 1MUP, which is most similar to uMUP-II) as a template (Bocskei et al., 1992). The model was constructed initially with the original thiazole ligand in place to ensure the disposition of side chain groups that were consistent with general ligand binding and to prevent cavity `collapse'. The model and original structure were then used for docking calculations using the program ICM (Abagyan and Totrov, 1994). The NPN ligand structure, originally constructed using WebLabViewer Pro (MSI), was inserted into the cavity by crude superposition onto thiazole. The thiazole was then removed, and a Monte Carlo search was run for 250,000 steps. The protein backbone was kept fixed although all side chain groups around the cavity (22 groups, Figure. 7 ▶) and the ligand were unconstrained. In both cases, the simulations converged to a solution well within the full course of the protocol, and the lowest energy conformers were saved as the predicted binding conformations. Binding energy estimates were calculated from the total energy δE:
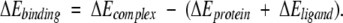 |
2 |
Acknowledgments
This work was supported by grants from the BBSRC to R.J.B. (SO6165) and J.L.H. (SO5935, FDP/93/30). A.D.M. gratefully acknowledges the Egyptian Government for a postgraduate studentship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.31701.
References
- Abagyan, R.A. and Totrov, M.M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235 983–1102. [DOI] [PubMed] [Google Scholar]
- Bacchini, A., Gaetani, E., and Cavaggioni, A. 1992. Pheromone binding proteins of the mouse, Mus musculus. Experientia 48 419–421. [DOI] [PubMed] [Google Scholar]
- Beynon, R.J., Robertson, D.H.L., Hubbard, S.J., Gaskell, S.J., and Hurst, J.L. 1999. The role of protein binding in chemical communication: Major urinary proteins in the house mouse. In Advances in chemical communication in vertebrates (eds. R.E. Johnston et al.), pp. 137–147. Plenum Press, New York.
- Bocskei, Z., Groom, C.R., Flower, D.R., Wright, C.E., Phillips, S.E.V., Cavaggioni, A., Findlay, J.B.C., and North, A.C.T. 1992. Pheromone binding to two rodent urinary proteins revealed by X-ray crystallography. Nature 360 186–188. [DOI] [PubMed] [Google Scholar]
- Brennan, P.A., Schellinck, H.M., and Keverne, E.B. 1999. Patterns of expression of the immediate-early gene egr-1 in the accessory olfactory bulb of female mice exposed to pheromonal constituents of male urine. Neuroscience 90 1463–1470. [DOI] [PubMed] [Google Scholar]
- Brito, R.M. and Vaz, W.L. 1986. Determination of the critical micelle concentration of surfactants using the fluorescent probe N-phenyl-1-naphthylamine. Anal. Biochem. 152 250–255. [DOI] [PubMed] [Google Scholar]
- Cavaggioni, A., Mucignat, C., and Tirindelli, R. 1999. Pheromone signalling in the mouse: Role of urinary proteins and vomeronasal organ. Arch. Ital. Biol. 137 193–200. [PubMed] [Google Scholar]
- Epps, D.E., Raub, T.J., and Kedzy, F.J. 1995. A general, wide-range spectrofluorometric method for measuring the site-specific affinities of drugs toward human serum albumin. Anal. Biochem. 227 342–350. [DOI] [PubMed] [Google Scholar]
- Flower, D.R. 1996. The lipocalin protein family: Structure and function. Biochem. J. 318 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries, R.E., Robertson, D.H.L., Beynon, R.J., and Hurst, J.L. 1999. Unravelling the chemical basis of competitive scent marking in house mice. Anim. Behav. 58 1177–1190. [DOI] [PubMed] [Google Scholar]
- Hurst, J.L., Robertson, D.H.L., Tolladay, U., and Beynon, R.J. 1998. Proteins in urine scent marks of male house mice extend the longevity of olfactory signals. Anim. Behav. 55 1289–1297. [DOI] [PubMed] [Google Scholar]
- Krieger, J., Schmitt, A., Lobel, D., Gudermann, T., Schultz, G., Breer, H., and Boekhoff, I. 1999. Selective activation of G protein subtypes in the vomeronasal organ upon stimulation with urine-derived compounds. J. Biol. Chem. 274 4655–4662. [DOI] [PubMed] [Google Scholar]
- Mucignat-Caretta, C., Caretta, A., and Cavaggioni, A. 1995. Acceleration of puberty onset in female mice by male urinary proteins. J. Physiol. 486 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocaktan, A., Yoneyama, H., and Nakae, T. 1997. Use of fluorescence probes to monitor function of the subunit proteins of the MexA-MexB-oprM drug extrusion machinery in Pseudomonas aeruginosa. J. Biol. Chem. 272 21964–21969. [DOI] [PubMed] [Google Scholar]
- Pes, D., Robertson, D.H.L., Hurst, J.L., Gaskell, S.J., and Beynon, R.J. 1999. How many major urinary proteins are produced by the house mouse Mus domesticus? In: Advances in chemical communication in vertebrates. (eds. R.E. Johnston et al.) pp. 149–161. Plenum Press, New York.
- Robertson, D.H.L., Beynon, R.J., and Evershed, R.P. 1993. Extraction, characterization and binding analysis of two pheromonally active ligands associated with major urinary protein of house mouse (Mus musculus). J. Chem. Ecol. 19 1405–1416. [DOI] [PubMed] [Google Scholar]
- Robertson, D.H.L., Cox, K.A., Gaskell, S.J., Evershed, R.P., and Beynon, R.J. 1996. Molecular heterogeneity in the major urinary proteins of the house mouse Mus musculus. Biochem. J. 316 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, D.H.L., Hurst, J.L., Bolgar, M.S., Gaskell, S.J., and Beynon, R.J. 1997. Molecular heterogeneity of urinary proteins in wild house mouse populations. Rapid Commun. Mass Spectrom. 11 786–790. [DOI] [PubMed] [Google Scholar]
- Sali, A. and Blundell, T.L. 1993. Comparative protein model building by satisfaction of spatial restraints. J. Mol. Biol. 234 779–815. [DOI] [PubMed] [Google Scholar]
- Zidek, L., Stone, M.J., Lato, S.M., Pagel, M.D., Miao, Z., Ellington, A.D., and Novotny, M.V. 1999. NMR mapping of the recombinant mouse major urinary protein I binding site occupied by the pheromone 2-sec-butyl-4,5-dihydrothiazole. Biochemistry 38 9850–9861. [DOI] [PubMed] [Google Scholar]