

Abstract
It is widely accepted that solvent-exposed sites in proteins play only a neglible role in determining protein energetics. In this paper we show that amino acid substitutions at the fully exposed Lys15 in bovine pancreatic trypsin inhibitor (BPTI) influenced the CD- and DSC-monitored stability: The Tden difference between the least (P1 Trp) and the most stable (P1 His) mutant is 11.2°C at pH 2.0. The ΔHden versus Tden plot for all the variants at three pH values (2.0, 2.5, 3.0) is linear (ΔCp,den = 0.41 kcal• mole−1 • K−1; 1 cal = 4.18 J) leading to a ΔGden difference of 2.1 kcal•mole−1. Thermal denaturation of the variants monitored by CD signal at pH 2.0 in the presence of 6 M GdmCl again showed differences in their stability, albeit somewhat smaller (ΔTden =7.1°C). Selective reduction of the Cys14–Cys 38 disulfide bond, which is located in the vicinity of the P1 position did not eliminate the stability differences. A correlation analysis of the P1 stability with different properties of amino acids suggests that two mechanisms may be responsible for the observed stability differences: the reverse hydrophobic effect and amino acid propensities to occur in nonoptimal dihedral angles adopted by the P1 position. The former effect operates at the denatured state level and causes a drop in protein stability for hydrophobic side chains, due to their decreased exposure upon denaturation. The latter factor influences the native state energetics and results from intrinsic properties of amino acids in a way similar to those observed for secondary structure propensities. In conclusion, our results suggest that the protein-stability-derived secondary structure propensity scales should be taken with more caution.
Keywords: Thermodynamic stability, solvent-exposed residue, reverse hydrophobic effect, bovine pancreatic trypsin inhibitor
It is generally accepted that mutations that affect stability occur at sites of low solvent accessibility and of low crystallographic thermal factors. This suggests that buried, well-defined interactions make a larger contibution to the global stability than solvent-exposed side chains (Alber et al. 1987). This is in agreement with the general observation that internal amino acid residues have higher evolutionary conservation tendency than those located in exposed regions (e.g., localized in surface loops). The precise packing of protein hydrophobic core dictated by docking of secondary structure segments appears to be the major determinant of protein stability. Creation of cavities and placing charged residues in the protein interior significantly lowers protein stability (Matthews 1996). Conversely, the protein surface is more hydrophilic, shows a partly charged character, and is considered to play a passive role in protein folding and stability. This role has not been investigated much. It may be, however, expected that amino acid substitution on the protein surface will marginally influence protein stability because no change in exposure to solvent occurs in the native and denatured state of wild-type and mutant protein.
There are different types of interactions that stabilize the native protein structure: hydrogen bonds, electrostatic interactions, and van der Waals forces (Matthews 1996; Pace et al. 1996). An important energetic role is also played by various entropic contributions, particularly the conformational and hydration entropies. Factors that determine protein thermodynamic stability have been studied extensively through mutational analysis, but few general rules have emerged that are able to predict quantitatively the stability effect of a single mutation. The energetic role played by a given amino acid residue results from the differences in the side-chain exposure to the solvent, the rigidity/flexibility of the local side-chain environment, the location in different types of secondary structure, and the highly cooperative nature of protein structure. Moreover, it has been suggested that residual structure in the protein denatured state may also influence the effect of mutation (Green et al. 1992).
The model protein used in this study is bovine pancreatic trypsin inhibitor (BPTI), a small monomeric globular protein of 58 amino acids with three disulfide bonds, containing both α-helix and β-sheet, and a defined hydrophobic core. BPTI is stable even in 6 M GdmCl and shows very high thermal stability, its denaturation temperature at neutral pH exceeds 100°C (Moses and Hinz 1983; Makhatadze et al. 1993). The focus of this study is on Lys15 (the P1 site in the nomenclature of Schechter and Berger 1967), the residue which is fully solvent exposed and is a major determinant of the energetics and specificity of proteinase recognition (Krowarsch et al. 1999). To probe the role of the P1 residue on the protein thermodynamic stability, Lys15 was mutated to 17 different amino acids and the thermodynamic stabilities of the variants were determined using CD and calorimetric measurements.
Results
The P1 position
Figure 1 ▶ shows a schematic view of the crystal structure of wild-type BPTI. The P1 residue (Lys15) is located in the central part of the proteinase binding loop, a six-residue (P3-P3′) segment of a convex shape and conserved, canonical conformation in many structurally distinct families of serine proteinase protein inhibitors (Bode and Huber 1992; Apostoluk and Otlewski 1998). The backbone dihedral angles of Lys15 in the free state of the wild-type inhibitor are similar in different crystal forms (Deisenhofer and Steigemann 1975; Wlodawer et al. 1984, 1987) and also in the high resolution solution NMR structure (Berndt et al. 1992; averaged values: φ = −104 ± 10°, ψ = 24 ±10°). The P1 main-chain angles lie in the bridge between the α-helical and β-sheet regions of the Ramachandran map and are most similar to position i + 2 of type I β-turn (φ = −91°, ψ= −7° Hutchinson and Thornton 1994). Although no spatial structures of free BPTI with amino acids other than Lys or Arg (Czapinska et al. 2000) at P1 have been determined, our study of the complexes between ten different P1 variants of BPTI and trypsin shows that in the complexed state the main-chain dihedral angles are insensitive to mutation and are very similar to the wild type (Helland et al. 1999). The Lys15 side chain is 94% solvent exposed when compared with the accessible surface area of a Lys residue in an extended Ala-Xaa-Ala tripeptide (Richards 1977). It is, therefore, reasonable to assume that mutation of Lys15 to other amino acids should have no effect on the overall protein stability.
Fig. 1.

(A) Schematic representation of the crystal structure of BPTI (Wlodawer et al. 1987). Solvent-exposed Lys15 side chain is shown in the central part of the canonical-binding loop together with three disulfide bonds and secondary structure elements. (B) Superimposition of the main-chain conformation of the protease-binding loop (P3–P3′ segment) of BPTI (PDB code 5pti), SSI (3ssi), and OMTKY3 (2ovo). (C) The Ramachandran map showing ϕ, ψ angles adopted by the surface-exposed residues of several proteins that are mentoned in the text.
Effect of P1 substitution on the native conformation
The first step involved mutating Lys15 to 17 different amino acids. The P1 Pro and Cys variants could not be produced in sufficient quantities, as we observed accumulation of many single- and double-disulfide intermediates and very little native protein during reoxidation of the reduced protein monitored by reversed-phase HPLC.
We applied circular dichroism spectroscopy as a sensitive probe of the overall conformation of the mutants in their native state. CD spectra of all P1 variants recorded at pH 2.0 were very similar in shape and intensity to the wild-type protein (Fig. 2 ▶). The molar ellipticity value of −5.7 × 105 deg•cm−1•mole−1 at 222 nm indicates the secondary structure integrity and the ellipticity at 274 nm shows the preservation of tertiary interactions in all mutants (the spectra of P1 Phe, Tyr, and Trp variants showed minor differences). For Trp15 BPTI, fluorescence emission spectrum showed a maximum at 354 nm, which is typical for a fully exposed indole ring (data not shown).
Fig. 2.

Circular dichroism spectra of P1 variants of BPTI in 10 mM formate at pH 2.0 at 298 K. The scans were recorded at 50 nm•min−1 with a step resolution of 1 nm. Each spectrum is the average of five scans. (Left) Spectra recorded in the 195–240 nm range (protein concentration = 3 × 10−4 M, 1-mm cuvette). (Right) Spectra recorded in the 240–340 nm range (protein concentration = 3 × 10−5 M, 10-mm cuvette).
Thermal denaturation
Typical denaturation curves of two P1 variants followed with DSC are shown in Figure 3 ▶. Routinely we performed DSC scans up to 125°C. Under these conditions denaturation is ∼70% reversible at pH 2.0 and ∼60% reversible at pH 3.0. Higher reversibility (∼90%) was observed when the CD signal was monitored at pH 2.0 with temperatures not exceeding 100°C. In agreement, Kim et al. (1993) showed that at high temperatures and pH>3.5 BPTI undergoes hydrolysis of two peptide bonds: Pro2-Asn and Asn3-Phe. Similar values of calorimetric and van't Hoff enthalpies (Table 1) and the agreement of the calculated lines with the experimental heat peaks (Fig. 3 ▶) indicate that the unfolding reaction is well approximated by a two-state model, similarly as for many other single-domain globular proteins.
Fig. 3.

Temperature dependence of the partial molar heat capacity of the least and most stable P1 mutants of BPTI : P1 Trp (189 μg) (A) and P1 His (193 μg) (B) variant of BPTI. DSC scans were performed in 10 mM glycine-HCl at pH 2.0. The best approximation using a two-state is depicted by a continuous line.
Table 1.
Thermodynamic parameters derived from DSC measurements of denaturation of P1 variants of BPTI determined in 10 mM glycine-HCl buffers
| P1 | pH | Tden (°C) | ΔHcal (kcal • mole−1) | ΔHvH (kcal • mole−1) | ΔHcal/ΔHvH | ΔCp,den (kcal • K−1•mole−1 | Concn. (μg/mL) | ΔGdenb (kcal • mole−1) |
| Trp | 2.0 | 75.6 | 65.12 | 65.84 | 0.99 | 0.22 | 200 | −1.03 |
| 2.5 | 78.8 | 66.00 | 69.00 | 0.99 | 0.43 | 120 | −0.85 | |
| Ile | 2.0 | 76.5 | 65.12 | 67.61 | 0.96 | 0.36 | 140 | −0.85 |
| 2.5 | 79.8 | 67.49 | 69.28 | 0.97 | 0.37 | 250 | −0.65 | |
| 3.0 | 85.6 | 68.54 | 70.23 | 0.97 | 0.43 | 120 | −0.69 | |
| Phe | 2.0 | 77.2 | 65.24 | 67.61 | 0.96 | 0.24 | 140 | −0.72 |
| 2.5 | 80.2 | 66.24 | 67.61 | 0.98 | 0.55 | 90 | −0.56 | |
| 3.0 | 85.8 | 68.80 | 74.30 | 0.93 | 0.43 | 150 | −0.69 | |
| Tyr | 2.0 | 77.3 | 65.90 | 64.32 | 1.02 | 0.40 | 300 | −0.70 |
| 2.5 | 80.5 | 67.20 | 66.20 | 1.02 | 0.36 | 130 | −0.49 | |
| 3.0 | 85.1 | 70.06 | 69.30 | 1.01 | 0.40 | 120 | −0.78 | |
| Leu | 2.0 | 77.7 | 66.40 | 63.30 | 1.05 | 0.33 | 80 | −0.63 |
| 2.5 | 81.7 | 67.90 | 69.00 | 0.98 | 0.25 | 50 | −0.28 | |
| 3.0 | 87.0 | 69.52 | 73.10 | 0.95 | 0.27 | 60 | −0.43 | |
| Val | 2.0 | 77.9 | 66.91 | 68.56 | 0.97 | 0.29 | 180 | −0.60 |
| 2.5 | 80.3 | 68.20 | 70.71 | 0.96 | 0.57 | 120 | −0.57 | |
| 3.0 | 85.4 | 70.02 | 73.58 | 0.95 | 0.41 | 140 | −0.77 | |
| Met | 2.0 | 78.8 | 66.00 | 63.80 | 1.03 | 0.40 | 130 | −0.42 |
| 2.5 | 80.0 | 67.90 | 70.70 | 0.96 | 0.31 | 60 | −0.63 | |
| 3.0 | 86.8 | 70.00 | 69.90 | 1.00 | 0.32 | 90 | −0.45 | |
| Glu | 2.0 | 77.2 | 71.70 | 79.10 | 0.94 | 0.20 | 250 | −0.36 |
| Gln | 2.0 | 79.5 | 66.50 | 65.20 | 1.02 | 0.32 | 200 | −0.28 |
| 2.5 | 82.8 | 68.10 | 68.70 | 0.99 | 0.23 | 230 | −0.06 | |
| 3.0 | 88.2 | 70.20 | 67.00 | 1.05 | 0.36 | 250 | −0.18 | |
| Ala | 2.0 | 79.6 | 67.30 | 66.30 | 1.02 | 0.42 | 100 | −0.27 |
| Thr | 2.0 | 80.8 | 67.50 | 70.40 | 0.96 | 0.24 | 200 | −0.04 |
| 2.5 | 84.4 | 68.70 | 66.00 | 1.04 | 0.30 | 220 | 0.23 | |
| 3.0 | 90.6 | 73.20 | 73.30 | 1.00 | 0.26 | 120 | 0.29 | |
| Ser | 2.0 | 81.4 | 68.49 | 69.71 | 0.98 | 0.45 | 210 | 0.08 |
| 2.5 | 84.6 | 68.99 | 70.00 | 0.98 | 0.43 | 120 | 0.29 | |
| 3.0 | 90.4 | 72.15 | 76.45 | 0.94 | 0.41 | 120 | 0.27 | |
| Gly | 2.0 | 81.5 | 67.75 | 68.25 | 0.99 | 0.24 | 260 | 0.09 |
| 2.5 | 84.7 | 68.25 | 68.56 | 0.99 | 0.29 | 280 | 0.30 | |
| 3.0 | 89.8 | 70.95 | 72.15 | 0.98 | 0.19 | 220 | 0.13 | |
| Asp | 2.0 | 82.0 | 67.85 | 70.71 | 0.96 | 0.33 | 230 | 0.19 |
| 2.5 | 84.4 | 70.20 | 76.00 | 0.92 | 0.28 | 200 | 0.27 | |
| 3.0 | 89.4 | 71.20 | 76.00 | 0.94 | 0.36 | 120 | 0.06 | |
| Asn | 2.0 | 82.3 | 67.99 | 70.47 | 0.96 | 0.36 | 200 | 0.25 |
| 2.5 | 84.9 | 69.52 | 72.38 | 0.96 | 0.43 | 200 | 0.36 | |
| Lysa | 2.0 | 83.3 | 68.00 | 63.00 | 1.08 | 0.22 | 110 | 0.44 |
| 2.5 | 86.3 | 70.00 | 67.80 | 1.03 | 0.3 | 90 | 0.60 | |
| 3.0 | 91.7 | 71.00 | 69.80 | 1.02 | 0.35 | 120 | 0.49 | |
| Arg | 2.0 | 83.6 | 68.44 | 67.85 | 1.00 | 0.31 | 270 | 0.45 |
| 2.5 | 87.3 | 70.47 | 73.58 | 0.96 | 0.41 | 250 | 0.85 | |
| 3.0 | 93.3 | 72.29 | 76.21 | 0.95 | 0.33 | 300 | 0.87 | |
| His | 2.0 | 86.7 | 69.90 | 69.28 | 1.00 | 0.33 | 180 | 1.09 |
| 2.5 | 90.4 | 71.74 | 74.30 | 0.96 | 0.29 | 150 | 1.48 | |
| 3.0 | 95.5 | 73.27 | 75.49 | 0.97 | 0.33 | 180 | 1.30 |
a Recombinant Lys15, Met52Leu BPTI.
b Free energy change of unfolding were calculated at 81°C, 83°C, and 89°C (the mean Tden values of P1 variants at pH 2.0, 2.5, and 3.0, respectively).
Figure 3 ▶ and Table 1 show that the Tden values for P1 variants are not identical but show substantial differences. At pH 2.0 the Tden values for the most (P1 His) and least (P1 Trp) stable variant differ by 11.1°C (Fig. 3 ▶). ΔHcal versus Tden data points fall on the same straight line for all the mutants (Fig. 4 ▶) implicating the same value of ΔCp,den and indicating that the differences in Tden values translate directly into differences in ΔGden values. At pH 2.0 the range of ΔGden value approaches 2.1 kcal•mole−1 (i.e., ∼20% of ΔGden at 25°C; Table1).
Fig. 4.

Temperature dependence of calorimetric enthalpy for 18 P1 variants of BPTI determined in 10 mM glycine-HCl buffers at pH 2.0, 2.5, and 3.0 (slope = 0.41 kcal•K−1•mole−1).
Stability of P1 variants at pH 2.0 in the presence of 6 M GdmCl
Moses and Hinz (1983) reported that in 6 M GdmCl at pH 2.0 wild-type BPTI has Tden of ∼65°C. Taking advantage of this exceptional thermodynamic stability, we performed CD thermal measurements of eight P1 variants in the presence of 6 M GdmCl. We observed clear unfolding transitions, similar to those in the absence of the denaturant, but shifted down by ∼22°C (Fig. 5 ▶). The ranking of stabilities correlates well with that in the absence of GdmCl (Fig. 6 ▶), but in 6 M GdmCl smaller dynamic range of Tden values was found (7.1°C versus 11.1°C), if the same sets of variants were compared (Table 2).
Fig. 5.

Thermal unfolding transitions curves of P1 Trp, P1 Met and P1 His BPTI variants in 10 mM glycine-HCl at pH 2.0, containing 6 M GdmCl, followed by the residual ellipticity at 222 nm.
Fig. 6.

Correlation plot of Tden values of P1 variants of BPTI measured in 10 mM glycine-HCl, pH 2.0 versus Tden values measured in the same buffer containing 6 M GdmCl.
Table 2.
Thermodynamic denaturation parameters for temperature unfolding of P1 variants of BPTI determined in 10 mM glycine-HCl, 6 M GdmCl, pH 2.0
| P1 | Tden (°C) | ΔHvH (kcal • mole−1) |
| Trp | 54.4 | 40.4 |
| His | 61.5 | 41.0 |
| Arg | 59.2 | 44.3 |
| Asp | 61.4 | 46.5 |
| Phe | 56.3 | 41.7 |
| Lys | 58.9 | 45.0 |
| Met | 58.8 | 45.1 |
| Thr | 60.1 | 44.3 |
Effect of selective reduction of the Cys14–Cys38 disulfide bond
BPTI contains three disulfide bonds: the Cys5–Cys55 and Cys30–Cys51 disulfides are buried and the Cys14–Cys38 one is exposed to a solvent that enables its selective reduction. To reduce the possible strain on the reactive-site binding loop and main-chain dihedral angles of the P1 residue, we selectively reduced this crosslink in five P1 variants using sodium borohydride. Then we measured their thermal stabilities by CD spectroscopy (Fig. 7 ▶). The selective reduction of the Cys14–Cys38 disulfide lowers Tden for all assayed variants by ∼25°C (Table 3). The ranking of stability differences remains similar to that for the fully oxidized variants. The correlation of Tden values for oxidized versus selectively reduced variants gives r = 0.75 and s = 0.45 (Table 4). When the data point for P1 Val, which is clearly off the correlation, is eliminated the r value increases to 0.98 and the s increases to 0.61.
Fig. 7.

Thermal unfolding transitions of Cys14–Cys38–reduced P1 Trp, P1 Arg, and P1 His BPTI variants in 10 mM glycine-HCl at pH 2.0. The transition curves were monitored by the residual ellipticity at 222 nm.
Table 3.
Thermodynamic denaturation parameters for temperature unfolding of Cys14–Cys38 reduced P1 variants of BPTI determined in 10 mM glycine-HCl, pH 2.0
| P1 | Tden (°C) | ΔHvH (kcal • mole−1) |
| Trp | 51.2 | 45.7 |
| Thr | 53.2 | 44.5 |
| Arg | 55.7 | 48.0 |
| Val | 56.2 | 48.5 |
| His | 57.9 | 46.0 |
Table 4.
The values of r and s calculated for the correlation of ΔGden values of P1 variants of BPTI with literature data
| All points | Removed points | |||||||
| y-axis | # | r | s | aa | # | r | s | Reference |
| P1 BPTI GdmCl | 8 | 0.84 | 0.56 | Tden vs. Tden correlation (Fig. 6 ▶) | ||||
| P1 BPTI reduced | 5 | 0.75 | 0.45 | Val | 4 | 0.98 | 0.61 | Tden vs. Tden correlation (Fig. 8 ▶) |
| SSI | 8 | 0.82 | 1.31 | Gly | 7 | 0.97 | 1.58 | Tamura and Sturtevant (1995) |
| Rop | 18 | 0.53 | 0.75 | altered contents points | 9 | 0.62 | 1.03 | Predki et al. (1996) |
| OMTKY3 | 17 | 0.63 | 0.31 | His, Lys, Arg | 14 | 0.97 | 0.61 | Otlewski and Laskowski (1985) |
| λ Cro | 8 | 0.78 | 1.1 | His | 7 | 0.95 | 2.37 | Pakula and Sauer (1990) |
| cytochrome c | 11 | 0.78 | 2.37 | Leu | 10 | 0.92 | 0.91 | Herrmann et al. (1995) |
| Bowler et al. (1993) | ||||||||
| hydrophobicity | 18 | −0.87 | −1.49 | His | 17 | −0.91 | −2.12 | Fauchere and Pliska (1983) |
| hydrophobicity | 17 | −0.70 | −1.76 | His | 16 | −0.87 | −2.7 | Eisenberg and McLachlan (1986) |
| β-turn type I, residue i + 2 | 18 | 0.37 | 0.37 | statistically significant points | 10 | 0.87 | 1.66 | Hutchinson and Thornton (1994) |
| SSIa/β-turn type I, residue i + 2 | 8 | 0.85 | 0.78 | statistically significant points | 6 | 0.95 | 1.03 | Tamura and Sturtevant (1995) |
| Hutchinston and Thornton (1994) | ||||||||
| OMTKY3a/β-turn type I, residue i + 2 | 17 | 0.68 | 1.37 | statistically significant points | 9 | 0.86 | 1.98 | Otlewski and Laskowski (1985) |
| Hutchinson and Thornton (1994) | ||||||||
x-Axis contains ΔGden values of P1 mutants of BPTI (the last column of Table 1).
a x-Axis.
Discussion
There are numerous protein-stability data concerning the effect of the substitution of all (or almost all) amino acids at a single, solvent-exposed site. These data can be divided into two categories, depending on the local conformation of the main chain at the site. One line of experiments addresses the question of intrinsic tendency of an amino acid to occur in a particular type of secondary structure: α-helix (O'Neil and DeGrado 1990; Horovitz et al. 1992; Blaber et al. 1993; Myers et al. 1997), β-sheet (Kim and Berg, 1993; Minor and Kim 1994; Smith et al. 1994; Otzen and Fersht 1995), and β-turns (Predki et al. 1996). To minimize interactions with the protein structure and to generate clean propensity scales, such investigations are conducted at surface-exposed sites. The main conclusion is that several properties of the side chain contribute to the observed propensity scale, including steric clashes with the backbone, entropy loss, burial of nonpolar surface, and electrostatic interactions. Very important for the following discussion is that all these factors operate on the native state level. In principle, there is no need to explain the helix propensity in terms of possible effect(s) on the denatured state. A nice example was given by Myers et al. (1997), who showed that the helix propensity determined from stability experiments on a surface exposed site in ribonuclease T1 is the same as that determined from CD measurements of a model helical peptide of identical sequence.
The second line of studies does not refer to the location of the mutated site in a specific secondary structure element, rather the site is chosen exclusively to fulfill the condition of high exposure to a solvent (Pakula and Sauer 1990; Herrmann et al. 1995; Tamura and Sturtevant 1995; Smith et al. 1996). A characteristic feature of these stability studies is a reverse hydrophobic effect (i.e., a decrease of protein stability with increasing side-chain hydrophobicity, originally observed for the λ Cro protein (Pakula and Sauer 1990). It is postulated that the stability decrease results from weak interactions, which the introduced hydrophobic residue can form in the denatured state but due to full exposure, not in the native state. Such weak interactions are inherently difficult to observe directly. However, recent multidimensional NMR experiments show that the unfolded states of several proteins can contain a residual structure of either a nativelike or non-native-like character (Neri et al. 1992; Logan et al. 1994; Yi et al. 2000). Also detailed CD studies were able to detect residual structure in a barnase inhibitor–barstar (Nölting et al. 1997). Further, the presence of weak interactions in the denatured state was anticipated from the decrease of the m (Wrabl and Shortle 1999) and ΔCp,den (Herrmann and Bowler 1997) value, which was approximately proportional to the amount of new surface exposed upon the breakdown of the native state to the denatured state. Thus, reasoning in terms of either the native state, the denatured state, or both (Smith et al., 1996) is currently used to explain the differences in stability at solvent-exposed sites in proteins. Because the whole range of stability between the most and the least stabilizing amino acid in all above mentioned analyses is typically only –1–2 kcal•mole−1, a clear explanation in thermodynamic and structural terms remains difficult.
The effect observed in this study—2.1 kcal•mole−1—is similar in range to those mentioned above. The whole range of stabilities is comparable to the destabilization caused removing of about two methylene groups from the protein interior (Matthews 1996). To reveal origin of the effect, we applied the Pearson-product moment correlation analysis of our ΔGden values with 245 different properties of amino acid side chains reported by Nakai et al. (1988) and available protein-stability data on surface-exposed sites. The correlation provides two values: r, the correlation coefficient, and s, the slope (power dependence) of the correlation. Some of the most prominent correlations are reported in Table 4. It should be mentioned that these correlations were performed for different numbers of data points and that the stability data were determined under different conditions (particularly pH). pH Difference can affect comparisons for ionizing side chains. In several cases, visual inspection showed that one or two data points were clearly off the plot. Their elimination provided a much better correlation coefficient and is included in Table 4.
There are indications showing that the observed stability differences at the P1 position of BPTI can be due to residual hydrophobic interactions in the denatured state. The P1 stability differences correlate well with the hydrophobicity scales of amino acid side chains (Table 4). For example, the correlation with the solvation free energy parameter (Eisenberg and McLachlan 1986) gave r = − 0.70, and r = −0.78 was obtained for the correlation with the scale of Fauchere and Pliska (1983). In both cases, elimination of the His point increased the r value to −0.87 and −0.91, respectively. These correlations suggest a mechanism to which the more hydrophobic side chains can more efficiently interact with the denatured state and, therefore, destabilize the protein. In effect, protein stability is negatively correlated with side-chain hydrophobicity. Our data show that removing a single methylene group from the protein surface (Ile→Val, Leu→Val, or Ala→Gly comparisons) stabilizes BPTI by ∼0.25–0.36 kcal•mole−1 (Table1), that is, ∼25% of the destabilizing effect of the same mutation performed in protein interior (Matthews 1996). This is also ∼25% of the effect measured for the favorable transfer of the P1 aliphatic side chains to S1 hydrophobic pockets of several proteinases (Lu et al. 1997; Krowarsch et al. 1999).
Clear correlations (the r value in the range of 0.7 to 0.9) with the hydrophobicity scales were also observed for substitutions at the surface exposed sites of four other proteins: Streptomyces subtilisin inhibitor (SSI; Tamura and Sturtevant 1995), turkey ovomucoid third domain (OMTKY3; Otlewski and Laskowski 1985), λ Cro (Pakula and Sauer 1990), iso-1-cytochrome c (Bowler et al. 1993; Herrmann et al. 1995). The investigators of these studies postulate that hydrophobic side chains, when introduced at the solvent-exposed sites, are more buried in the denatured state than in the native state, which results in a decreased stability. Our P1 stability effect in BPTI correlates with ΔGden values for the above mentioned proteins (Table 4). The correlation coefficient varies from 0.63 to 0.82, depending on the protein. This means that the rankings of amino acids stabilising the denatured state of different proteins (including BPTI) are not the same.
Further, this suggests that the reverse hydrophobic effect operates differently for the four proteins, which may result from the differences in the denaturation methods applied and, therefore, from the properties of their denatured state. Perhaps this is just a reminiscence of the differences in hydrophobicity scales of amino acids, which in the case of transfer-based evaluations depend on the hydrophobic phase used. On the other hand, it should be mentioned that BPTI/SSI and BPTI/λ Cro correlations are the two strongest ones of the 245 we have analyzed.
The origin and interpretation of the reverse hydrophobic effect may be related to a reference value of accessible surface area in the denatured state of protein. In this paper, it was calculated using an extended Ala-Xaa-Ala tripeptide model (Richards 1977). However, Creamer et al. (1995), using simulated hard-sphere-peptide and native-protein-fragment models, showed that the tripeptide-based calculations severely overestimate the side-chain accessible area in the denatured protein. Their calculations also show that reduction of the area is larger for longer peptides. According to this model, the accessible surface area of the P1 side chain in BPTI in the denatured state is smaller than expected from the tripeptide model, which explains the observed stability trend.
However, when thermal denaturation was repeated for eight BPTI P1 mutants in the presence of 6 M GdmCl, the stability rank remained essentially unchanged (the correlation coefficient r = 0.84), although the range of the Tden values decreased from 11.1°C to 7.1°C (Fig. 5 ▶, Table 2). This shows that ∼65% of the stability effect remained in the presence of 6 M GdmCl. The effect of 6 M GdmCl on protein conformation is a subject of a long-term debate (Matthews 1993; Shortle 1993). It is often considered as the most universal model for the fully unfolded state. However, the free energy of transfer from a nonpolar solvent to 6 M GdmCl is equal to ∼30% of transfer-free energy to water (Creighton 1979) suggesting that residual structure is highly probable. Nevertheless, it is difficult to imagine that weak hydrophobic interactions can persist in the denatured state in the presence of 6 M GdmCl and at elevated temperature, because 6 M GdmCl is commonly used to denature virtually all proteins at room temperature. In agreement, Privalov et al. (1989) argue that proteins in concentrated solutions of GdmCl attain a random-coil state at high temperature. In our opinion, this indicates that the reverse hydrophobic effect is not the only reason for the observed stability changes.
It is striking that three most stable variants contain positively charged residue (His, Arg, Lys) at the P1 site. These are also the residues that often weaken the correlations reported in Table 4. This suggests that the electrostatic effects either in the native or in the denatured state might be responsible for the observed stability effect. The possibility of the electrostatic stabilization in the native state seems to be unlikely as the BPTI molecule already contains several positively charged residues in the vicinity of the P1 position. Further, the rank of stabilities remains upon heating in 6 M GdmCl (Table 2), suggesting that electrostatic effects in the native and denatured state are unlikely.
Now we turn to the native state of BPTI to explain the observed stability differences. The conformation of the P3–P3′ segment, where the P1 position is centrally located, is similar in BPTI and in the SSI and OMTKY3 inhibitors. Figure 1 ▶ shows these segments in the three inhibitors and the distribution of their P1 main-chain angles in the Ramachandran map. As described in Results, the values of their ϕ and ψ angles are in a nonoptimal conformation, intermediate between α-helical and β-sheet (i + 2 position of β-turn I). We suppose that the energetics of this main-chain strain might depend on the amino acid side chain (i.e., there exists a propensity scale or potential for the i + 2 position of β-turn I). Thus, the correlations BPTI/SSI and BPTI/OMTKY3 may result not only from the reverse hydrophobic effect, but, alternatively, from the propensity scale of the amino acids to occur in this particular main-chain conformation.
To further investigate this possibility, we selectively reduced the Cys14–Cys38 disulfide bond in five P1 mutants with the aim to reduce strain on the main chain of the P1 position. The stability of these selectively reduced mutants differed by 6.7°C, compared to the 11.1°C difference found for the fully oxidized variants (Fig. 7 ▶, Table 3). Again, ∼60% of the total effect was insensitive to the selective reduction of the Cys14–Cys38 disulfide bond, suggesting that about half of the stability effect is related to the reverse hydrophobic effect and the other half can be explained in terms of native-state energetics.
Hutchinson and Thornton (1994) calculated PDB-based potentials for different positions in a number of β-turns, including the i + 2 position of turn I. Although our P1 stability data do not correlate (r = 0.37) with the position i + 2 of β-turn I potential (Table 4), the correlation becomes very good (r = 0.87) if only statistically significant (according to the investigators) data (10 data points) are used. Further, we also found a clear correlation with β-turn I potential for two other inhibitors: for OMTKY3, r = 0.68 (for the statistically significant data, r = 0.86) and for SSI, r = 0.85 (for the statistically significant data, r = 0.95). In a control search, we did not observe a correlation of our data with α-helical propensity scales of amino acids (the r value from 0 to −0.3, data not shown). There was also no correlation with the Rop mutants stability data (r = 0.53; Predki et al. 1996). The mutated residue 30 in Rop assumes completely different dihedral angles (Fig. 1 ▶).
In summary, there is no clear evidence providing a uniform mechanism for the observed stability changes at the exposed P1 site in BPTI. Two mechanisms may roughly contribute equally to the observed differences: the reverse hydrophobic effect, which operates on the denatured-state level, and different propensities of amino acids to occur in the nonoptimal dihedral angles of the P1 position, which operate on the native-state level. As the two mechanisms are fundamentally different and based on different features of amino acids, the P1 stability does not superbly correlate with properties of amino acids and available protein-stability data. We expect that both these phenomena play a significant role also in other stability studies. Further and more essential, the reverse hydrophobic effect may be the reason of relatively poor correlations observed among different protein-stability-derived propensity scales. The extent of the correlation may critically depend on the protein used, secondary structure, local conformation, and the denaturation method. For example, there are clear correlations (the r value from 0.6 to 0.79) between various β-sheet propensity and hydrophobicity scales, but the α-helix propensity/hydrophobicity correlations are very poor (the r ∼0.2–0.3; data not shown). Thus, the observed context dependence of β-sheet propensity scales (Minor and Kim 1994) might partially result from the reverse hydrophobic effect. In the case of α-helix, due to the well-defined main-chain angles and local (enthalpic and entropic) terms, the conformational, native-state effect dominates and the reverse hydrophobic effect provides only some noise to the propensity scales.
Materials and methods
Materials
GdmCl, urea, DMSO, DMF, methanol, acetonitrile, and the basic components of culture media were purchased from Merck (Germany). TFA and CNBr were obtained from Fluka (Switzerland). Tris, sodium acetate, DTT, chloramphenicol, ampicilin, GSH, and GSSG were from Sigma (USA). DNA-modifying enzymes (T4 polymerase, T4 DNA ligase, T4 polynucleotide kinase) were purchased from Boehringher (Germany). The DNA-sequencing kit was from Amersham (UK) and the DNA purification kit was from QIAGEN (USA). Oligonucleotides were chemically synthesized by Ransom Hill (USA). IPTG was from Bachem (Switzerland).
Expression and purification of BPTI mutants
All mutants of BPTI were overexpressed as fusion proteins in Escherichia coli strain BL21 (DE3) pLysS, using the T7 promotor system (Studier et al. 1990), as described by Staley and Kim (1994). The plasmids derived from pAED4 bearing a portion of the E. coli trp operon, which serves as a leader sequence, followed by a Met residue and the mutant BPTI-encoding sequences, were prepared by site-directed mutagenesis (Kunkel et al. 1987). All recombinant variants contained additional Met52→Leu mutation to enable CNBr cleavage of the fusion protein. Details of the purification protocol were published elsewhere (Krokoszynska et al. 1998).
Protein identity of all variants was confirmed using electrospray mass spectrometry with a Finnigan MAT TSQ-700 spectrometer equipped with an ESI source (mass agreement within ±1 Da).
Thermal-stability measurements
Differential scanning calorimetry (DSC) experiments were performed on a Nano II calorimeter (CSC Corp.). The experiments were done at protein concentration of 100–200 μg/mL (total cell volume: 323 μL) at the scan rate of 1.0 K•min−1 under pressure excess of 2.5 atm. Before the DSC run, the protein solution was extensively dialyzed against 10 mM glycine-HC at pH 2.0, 2.5, or 3.0. Repeated measurements with the same protein sample showed ∼70% reproducibility of ΔHcal values at pH 2.0. The partial specific volume of BPTI was assumed to be 0.71 cm3•g−1 (Makhatadze et al. 1993).
Tden, ΔHcal, ΔHvH, and ΔCp,den values were calculated from thermogram analysis using software CpCcalc provided by CSC Corp. Free-energy change of unfolding at 81°C (the mean Tden value of all P1 variants at pH 2.0) was calculated from the Gibbs-Helmholtz equation:
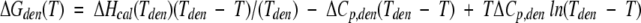 |
1 |
CD measurements
Spectra
CD spectra of the P1 variants were recorded in 10 mM formate at pH 2.0 on a Jasco J-715 spectropolarimeter. Measurements were made at a protein concentration of 3 × 10−5 M using a 10-mm cuvette (240–340 nm range) or at 3 × 10−4 M protein in a 1-mm pathlength cuvette (200–260 nm range).
Thermal transitions
Thermal denaturation was monitored following the ellipticity at 222 nm using band slit 2 nm and a response time of 4 sec. Automatic Peltier accessory PFD 350S allowed continuous monitoring of the thermal transition at a constant rate of 1 K • min−1. Temperature of the sample was monitored directly using a probe immersed in the cuvette and controlled with PFD-350S/350L Peltier type FDCD attachment.
Protein was dissolved in 10 mM glycine-HCl, pH 2.0 and passed through a 0.22-μm Millipore filter before measurement. In a series of experiments, the buffer additionally contained 6 M GdmCl. The data were analyzed assuming a two-state reversible equilibrium transition as follows:
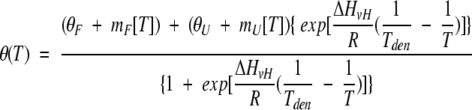 |
2 |
where T is the absolute temperature in K, R is the gas constant (1.98 cal•mole−1•K−1), θ(T) is the ellipticity signal at 222 nm, ΔHvH is the van't Hoff enthalpy, θF is the value of the folded signal extrapolated to 0 K, mF is the slope of the temperature dependence of the CD signal for the folded protein, θU is the value of the unfolded signal extrapolated to 0 K, mU is the slope of the temperature dependence of the CD signal for the unfolded protein, and Tden is the denaturation temperature.
Selective reduction of the Cys14–Cys38 disulfide bond
Selective reduction of the Cys14–Cys38 disulfide in several P1 variants of BPTI was performed according to Quast et al. (1975) using NaBH4 as the reducing agent. On average 2.1 ± 0.2 moles of −SH groups per mole of protein were found by the Ellman method (Ellman 1959) after desalting of reduced BPTI on a Sephadex G25 column equilibrated in carefully degassed 10 mM glycine-HCl at pH 2.0. Desalted samples were immediately used for thermal denaturation experiments. Repeated denaturation showed reversibility of the transition with a minor (5% of the total signal change) additional transition at ∼ 20°C higher temperature, most likely originating from partial oxidation of Cys14–Cys38 disulfide.
Fig. 8.

Correlation plot of Tden values of P1 variants of BPTI vs. Tden values of P1 variants with the Cys14–Cys38 bond reduced. Both data sets were determined in 10 mM glycine-HCl at pH 2.0.
Acknowledgments
The work was supported by grant 6 P04B 002 10 from the Polish Committee for Scientific Research. We thank Professor P.S. Kim for the plasmid bearing the wild-type BPTI gene; Lilia Joukova, Zofia Podgorska, and Marta Gladysz for excellent technical assistance; and Professor Zbigniew Szewczuk and Dr. Piotr Stefanowicz for mass-spectrometry experiments. We also appreciate the comments of anonymous referee 2.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
BPTI, bovine pancreatic trypsin inhibitor
OMTKY3, turkey ovomucoid third domain
SSI, Streptomyces subtilisin inhibitor
CD, circular dichroism
DSC, differential scanning calorimetry
GdmCl, guanidinium chloride
ΔHcal, calorimetric enthalpy
ΔHvH, van't Hoff enthalpy
ΔSden, ΔHden, ΔCp,den, and ΔGden, entropy, enthalpy, heat capacity, and free-energy changes, respectively, accompanying protein denaturation
Tden, denaturation temperature
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.38101.
References
- Alber, T., Dao-Pin, S., Nye, J.A., Muchmore, D.C., and Matthews, B.W. 1987. Temperature-sensitive mutations of bacteriophage T4 lysozyme occur at sites with low mobility and low solvent accessibility in the folded protein. Biochemistry 26 3754–3758. [DOI] [PubMed] [Google Scholar]
- Apostoluk, W. and Otlewski, J. 1998. Variability of the canonical loop conformations in serine proteinases inhibitors and other proteins. Proteins: Struct. Funct. Genet. 32 459–474. [PubMed] [Google Scholar]
- Berndt, K.D., Güntert, P., Orbons, L.P., and Wüthrich, K. 1992. Determination of a high quality nuclear magnetic resonance solution structure of the bovine pancreatic trypsin inhibitor and comparison with three crystal structures. J. Mol. Biol. 227 757–775. [DOI] [PubMed] [Google Scholar]
- Blaber, M., Zhang, X.J., and Matthews, B.W. 1993. Structural basis of amino acid alpha helix propensity. Science 260 1637–1640. [DOI] [PubMed] [Google Scholar]
- Bode, W. and Huber, R. 1992. Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204 433–451. [DOI] [PubMed] [Google Scholar]
- Bowler, B.E., May, K., Zaragoza, T., York, P., Dong, A., and Caughey, W.S. 1993. Destabilizing effects of replacing a surface lysine of cytochrome c with aromatic amino acids: Implications for the denatured state. Biochemistry 32 183–190. [DOI] [PubMed] [Google Scholar]
- Creamer, T.P., Srinivasan, R., and Rose, G.D. 1995. Modeling unfolded states of peptides and proteins. Biochemistry 34 16245–16250. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1979. Electrophoretic analysis of the unfolding of proteins by urea. J. Mol. Biol. 129 235–264. [DOI] [PubMed] [Google Scholar]
- Czapinska, H., Otlewski, J., Krzywda, S., Sheldrick, G.M., and Jaskolski, M. 2000. High resolution structure of bovine pancreatic trypsin inhibitor with altered binding loop sequence. J. Mol. Biol. 295 1237–1249. [DOI] [PubMed] [Google Scholar]
- Deisenhofer, J. and Steigemann, W. 1975. Crystallographic refinement of the structure of bovine pancreatic trypsin inhibitor at 1.5 å resolution. Acta Crystallogr. B31 238–250. [Google Scholar]
- Eisenberg, D. and McLachlan, A.D. 1986. Solvation energy in protein folding and binding. Nature 319 199–203. [DOI] [PubMed] [Google Scholar]
- Ellman, G.L. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82 70–77. [DOI] [PubMed] [Google Scholar]
- Fauchere, J.L. and Pliska. V. 1983. Hydrophobic parameters pi of amino-acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur. J. Med. Chem. 18 369–375. [Google Scholar]
- Green, S.M., Meeker, A.K., and Shortle, D. 1992. Contributions of the polar, uncharged amino acids to the stability of staphylococcal nuclease: Evidence for mutational effects on the free energy of the denatured state. Biochemistry 31 5717–5728. [DOI] [PubMed] [Google Scholar]
- Helland, R., Otlewski, J., Sundheim, O., Dadlez, M., and Smalas, A.O. 1999. The crystal structures of the complexes between bovine β-trypsin and ten P1 variants of BPTI. J. Mol. Biol. 287 923–942. [DOI] [PubMed] [Google Scholar]
- Herrmann, L.M. and Bowler, B.E. 1997. Thermal denaturation of iso-1-cytochrome c variants: Comparsion with solvent denaturation. Prot. Sci. 6 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann, L.M., Bowler, B.E., Dong, A., and Caughey, W.S. 1995. The effects of hydrophilic to hydrophobic surface mutations on the denatured state of iso-1-cytochrome c: Investigation of aliphatic residues. Biochemistry 34 3040–3047. [DOI] [PubMed] [Google Scholar]
- Horovitz, A., Matthews, J.M., and Fersht, A.R. 1992. α-Helix stability in proteins. II. Factors that influence stability at an internal position. J. Mol. Biol. 227 560–568. [DOI] [PubMed] [Google Scholar]
- Hutchinson, E.G. and Thronton, J.M. 1994. A revised set of potentials for β-turn formation in proteins. Prot. Sci. 3 2207–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, C.A. and Berg, J.M. 1993. Thermodynamic beta-sheet propensities measured using a zinc finger host peptide. Nature 362 267–270. [DOI] [PubMed] [Google Scholar]
- Kim, K.S., Tao, F., Fuchs, J., Danishefsky, A.T., Housset, D. Wlodawer, A., and Woodward, C. 1993. Crevice-forming mutants in the rigid core of bovine pancreatic trypsin inhibitor: Stability changes and new hydrophobic surface. Prot. Sci. 2 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokoszynska, I., Dadlez, M., and Otlewski, J. 1998. Structure of single-disulfide variants of bovine pancreatic trypsin inhibitor (BPTI) as probed by their binding to bovine β-trypsin. J. Mol. Biol. 275 503–513. [DOI] [PubMed] [Google Scholar]
- Krowarsch, D., Dadlez, M., Buczek, O., Krokoszynska, I., Smalas, A.O., and Otlewski J. 1999. Interscaffolding additivity: Binding of P1 variants of bovine pancreatic trypsin inhibitor to four serine proteases. J. Mol. Biol. 289 175–186. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A., Roberts, J.D., and Zakour, R.A. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Meth. Enzymol. 154 367–382. [DOI] [PubMed] [Google Scholar]
- Lu, W., Apostol, I., Qasim, M.A., Warne, N., Wynn, R., Zhang, W.L., Anderson, S., Chiang, Y.W., Ogin, E., Rothberg, I., et al. 1997. Binding of amino acid side-chains to S1 cavities of serine proteinases. J. Mol. Biol. 266 441–461. [DOI] [PubMed] [Google Scholar]
- Logan, T.M., Theriault, Y., and Fesik., S.W. 1994. Structural characterization of the FK 506 binding protein unfolded in urea and guanidine hydrochloride. J. Mol. Biol. 236 637–648. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G.I., Kim, K.S., Woodward, C., and Privalov, P.L. 1993. Thermodynamics of BPTI folding. Prot. Sci. 2 2028–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B.W. 1993. Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 62 139–160. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1996. Structural and genetic analysis of the folding and function of T4 lysozyme. FASEB J. 10 35–41. [DOI] [PubMed] [Google Scholar]
- Minor, D.L. and Kim, P.S. 1994. Measurement of the β-sheet-forming propensities of amino acids. Nature 367 660–663. [DOI] [PubMed] [Google Scholar]
- Moses, E. and Hinz, H.J. 1983. Basic pancreatic trypsin inhibitor has unusual thermodynamic stability parameters. J. Mol. Biol. 170 765–776. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1997. Helix propensities are identical in proteins and peptides. Biochemistry 36 10923–10929. [DOI] [PubMed] [Google Scholar]
- Nakai, K., Kidera, A., and Kanehisa, M. 1988. Cluster analysis of amino acid indices for prediction of protein structure and function. Prot. Eng. 2 93–100. [DOI] [PubMed] [Google Scholar]
- Neri, D., Billeter, M., Wider, G., and Wüthrich, K. 1992. NMR determination of residual structure in a urea-denatured protein, the 434-repressor. Science 257 1559–1563. [DOI] [PubMed] [Google Scholar]
- Nölting, B., Golbik, R., Soler-González, A., and Fersht, A.R. 1997. Circular dichroism of denatured barstar suggests residual structure. Biochemistry 36 9899–9905. [DOI] [PubMed] [Google Scholar]
- O'Neil, K.T. and DeGrado, W.F.A. 1990. A thermodynamic scale for the helix forming tendencies of the commonly occuring amino acids. Science 250 646–651. [DOI] [PubMed] [Google Scholar]
- Otlewski, J. and Laskowski Jr., M. 1985. Calorimetric investigation of the reactive site of turkey ovomucoid third domain. Fed. Proc. 44 1807. [Google Scholar]
- Otzen, D.E. and Fersht, A.R. 1995. Side-chain determinants of β-sheet stability. Biochemistry 34 5718–5724. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Shirley, B.A., McNutt, M., and Gajiwala, K. 1996. Forces contributing to the conformational stability of proteins. FASEB J. 10 75–83. [DOI] [PubMed] [Google Scholar]
- Pakula, A.A. and Sauer, R.T. 1990. Reverse hydrophobic effects relieved by amino-acid substitutions at a protein surface. Nature 344 363–364. [DOI] [PubMed] [Google Scholar]
- Predki, P.F., Agrawal, V., Brünger, A.T., and Regan, L. 1996. Amino-acid substitutions in a surface turn modulate protein stability. Nat. Struct. Biol. 3 54–58. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L., Tiktopulo, E.I., Venyaminov, S.Y., Griko, Y.V., Makhatadze, G.I., and Khechinashvili, N.N. 1989. Heat capacity and conformation of proteins in the denatured state. J. Mol. Biol. 205 737–750. [DOI] [PubMed] [Google Scholar]
- Quast, U., Engel, J., Steffen, E., Tschesche, H., Jering, H., and Kupfer, S. 1975. The effect of cleaving the reactive-site peptide bond Lys15-Ala16 on the conformation of bovine trypsin-kallikrein inhibitor (Kunitz) as revealed by solvent-perturbation spectra, circular dichroism and fluorescence. Eur. J. Biochem. 52 511–514. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. 1977. Areas, volumes, packing, and protein structure. Annu. Rev. Biophys. Bioeng. 6 151–176. [DOI] [PubMed] [Google Scholar]
- Schechter, I. and Berger, A. 1967. On the size of the active site in proteases. Biochem. Biophys. Res. Commun. 27 157–162. [DOI] [PubMed] [Google Scholar]
- Smith, C.K., Withka, J.M., and Regan, L. 1994. A thermodynamic scale for the β-sheet forming tendencies of the amino acids. Biochemistry 33 5510–5517. [DOI] [PubMed] [Google Scholar]
- Smith, C.K., Bu, Z., Anderson, K.S., Sturtevant, J.M., Engelman, D.M., and Regan. L. 1996. Surface point mutations that significantly alter the structure and stability of a protein's denatured state. Prot. Sci. 5 2009–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley, J.P. and Kim, P.S. 1994. Formation of a native-like subdomain in a partially folded intermediate of bovine pancreatic trypsin inhibitor. Prot. Sci. 3 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F., Rosenberg, A.H., Dunn, J.J., and Dubendorff, J.W. 1990. Use of T7 polymerase to direct expression of cloned genes. Meth. Enzymol. 185 60–89. [DOI] [PubMed] [Google Scholar]
- Tamura, A. and Sturtevant, J.M. 1995. A thermodynamic study of mutant forms of Streptomyces subtilisin inhibitor. III. Replacements of a hyper-exposed residue, Met73. J. Mol. Biol. 249 646–653. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Walter, J., Huber, R., and Sjolin, L. 1984. Structure of bovine pancreatic trypsin inhibitor. Results of joint neutron and X-ray refinement of crystal form II. J. Mol. Biol. 180 301–329. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Nachman, J., Gilliand, G.L., Gallagher, W., and Woodward, C. 1987. Structure of form III crystals of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 198 469–480. [DOI] [PubMed] [Google Scholar]
- Wrabl, J. and Shortle, D. 1999. A model of the changes in the denatured state structure underlying m value effect in staphylococcal nuclease. Nat. Struct. Biol. 6 876–883. [DOI] [PubMed] [Google Scholar]
- Yi, Q., Scalley-Kim, M.L., Alm, E.J., and Baker, D. 2000. NMR characterization of residual structure in the denatured state of protein L. J. Mol. Biol. 299 1341–1351. [DOI] [PubMed] [Google Scholar]