

Abstract
The catalytic center of an archaeal Type 2 RNase H has been identified by a combination of X-ray crystallographic and mutational analyses. The crystal structure of the Type 2 RNase H from Thermococcus kodakaraensis KOD1 has revealed that the N-terminal major domain adopts the RNase H fold, despite the poor sequence similarity to the Type 1 RNase H. Mutational analyses showed that the catalytic reaction requires four acidic residues, which are well conserved in the Type 1 RNase H and the members of the polynucleotidyl transferase family. Thus, the Type 1 and Type 2 RNases H seem to share a common catalytic mechanism, except for the requirement of histidine as a general base in the former enzyme. Combined with the results from deletion mutant analyses, the structure suggests that the C-terminal domain of the Type 2 RNase H is involved in the interaction with the DNA/RNA hybrid.
Keywords: Ribonuclease H, DNA/RNA hybrid, polynucleotidyl transferase family, crystal structure, mutational analysis
Ribonuclease H (RNase H) has a unique function to degrade specifically the RNA moiety of DNA/RNA hybrid duplexes (Hausen and Stein 1970). Biochemistry of this divalent metal-dependent endonuclease has been studied extensively because of its involvement in important biological processes such as DNA replication and repair (Kogoma and Foster 1998). Genes encoding RNase H have been found ubiquitously in viruses, bacteria, archaea, and eucarya, including human (Ohtani et al. 1999a). Because of the unique biochemical function, RNase H could be a potential target for medical applications. For example, RNase H is one of the therapeutic targets for AIDS (acquired immunodeficiency syndrome). The enzyme is also involved in antisense gene therapy.
RNase H is classified into two major types, according to their sequence similarity (Ohtani et al. 1999a, b). The biochemical and structural properties of the Type 1 RNase H, including the mechanisms of specific recognition of DNA/RNA hybrids and the catalytic reaction, have been studied extensively (Kanaya 1998; Morikawa and Katayanagi 1998). On the other hand, less information is available for the Type 2 enzyme, although a recent crystallographic study revealed the three-dimensional structure of an archaeal RNase HII (Lai et al. 2000). It would be interesting to understand how these two types of enzymes, which do not seem to share a common ancestor, catalyze the same hydrolytic reaction.
Haruki and coworkers (1998) previously cloned the rnhB gene encoding RNase HII (Tk-RNase HII) from a hyperthermophilic archaeon, Thermococcus kodakaraensis KOD1, and overexpressed the protein in Escherichia coli. The recombinant Tk-RNase HII, which will be simply designated as Tk-RNase HII hereafter, shows a relatively broad divalent cation specificity. This enzyme does not cleave the RNA-DNA junction itself, but cleaves to leave a single ribonucleotide at the 5′-end of the RNA-DNA junction.
We have determined the X-ray crystal structure of Tk-RNase HII. In combination with the characterization of mutant proteins, the crystal structure allows identification of the substrate recognition interface and the catalytic center.
Results and Discussion
Truncation of the protein for crystallization
Tk-RNase HII was crystallized initially by the vapor diffusion method using 5% PEG6000 as a precipitant. The crystal provided a diffraction pattern, which represents the primitive hexagonal lattice with unit cell dimensions of a = b= 62 (Å) and c = 115 (Å). Unfortunately, Bragg reflections were observed up to 3.5 Å resolution, which is insufficient for a structure determination at an atomic level.
In parallel with the optimization of the crystallization condition, limited proteolysis was examined to define the core fragments, which may yield a higher resolution crystal. Digestions of Tk-RNase HII with chymotrypsin and trypsin produced distinct fragments whose N-terminal sequences are the same as that of the full-length Tk-RNase HII, indicating preservation of the N-terminus in both fragments. The molecular mass of the chymotrypsin fragment was determined by MALDI-TOF mass spectroscopy to be 22,250 Daltons. Taking the preference of the protease into account, the fragment is presumed to range from residues 1–198. Similarly, the trypsin fragment, with a molecular mass of 24,488 Daltons, appeared to range from residues 1–217. Interestingly, the trypsin fragment retained nearly full activity, whereas the chymotrypsin fragment was almost completely inactive. These results suggest that the segment between 199 and 217 is crucial for the RNase H activity.
We have prepared five different truncated proteins for the crystallization trial: residues 1–198 (Tk-RNase HII-198), 1–203 (Tk-RNase HII-203), 1–207 (Tk-RNase HII-207), 1–213 (Tk-RNase HII-213), and 1–217 (Tk-RNase HII-217). Among them, Tk-RNase HII-213 produced rod-shaped crystals in a solution containing 15% (w/v) PEG6000 buffered by 200 mM MES at pH 6.5. The crystal, which belongs to the space group P212121 with unit cell dimensions of a = 43 (Å), b = 73 (Å), c = 78 (Å), diffracted to 2Å resolution with synchrotron radiation at the Photon Factory (Sakabe 1991). We attempted to determine the structure of this crystal form by the MIR method.
Overall structure
The folding of Tk-RNase HII-213 (the full-length molecule is composed of 228 amino acids) is shown in Figure 1 ▶. The structure is composed of two distinct domains. The N-terminal domain (residues 1–162) has a central core of a five-stranded β-sheet, in which only β2 is antiparallel to the other four. This sheet is surrounded by seven α-helices: α1–α7. As a whole, this domain with the α/β structure forms the well-known RNase H fold, which was found in the crystal structures of bacterial RNase HI (Katayanagi et al. 1990; Yang et al. 1990; Ishikawa et al. 1993), reverse transcriptase (Davies et al. 1991), integrase (Dyda et al. 1994; Bujacz et al. 1995), transposase (Rice and Mizuuchi 1995), and RuvC Holliday-junction resolvase (Ariyoshi et al. 1994). The C-terminal domain (residues 163–213) forms a compact structure consisting of two α-helices: α8 and α9. Unlike the N-terminal domain, bacterial RNases HI do not possess this domain. In the full-length enzyme, the α9 helix is followed by 15 amino acid residues (Fig. 2A ▶), of which conformation is likely to be flexible, as revealed from the susceptibility to trypsin digestion.
Fig. 1.

Stereodiagram showing the backbone structure of Tk-RNase HII-213. Helices, strands, and loops are shown in red, cyan, and orange, respectively. Green triangles with labeled amino acids indicate the sites in which the C-terminal truncations (Tk-RNase HII-198, Tk-RNase HII-203, Tk-RNase HII-207, and Tk-RNase HII-213) were constructed.
Fig. 2.
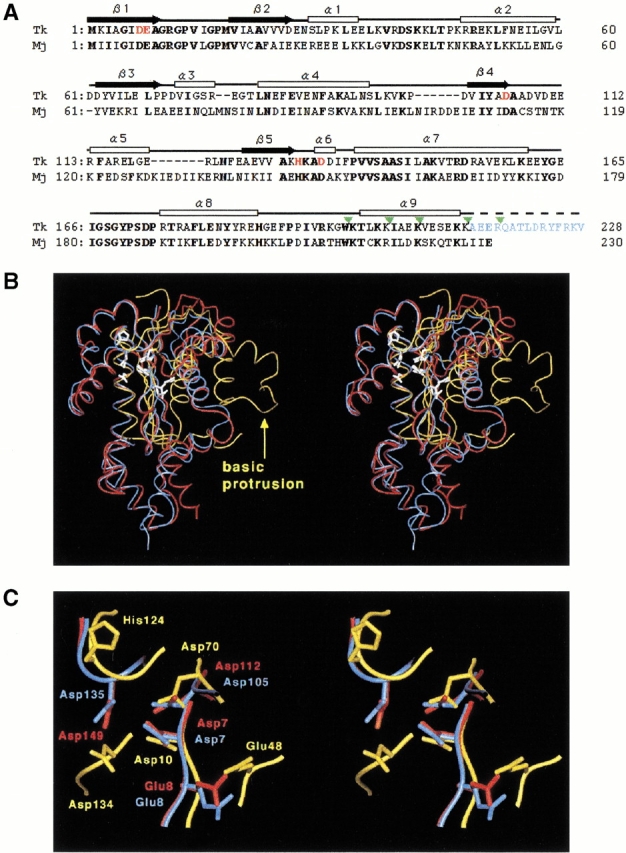
(A) Amino acid sequence alignment of Tk-RNase HII (Tk) and Mj-RNase HII (Mj) based on the comparison of their three-dimensional structures. The range of the secondary structures of Tk-RNase HII are shown above the sequence (white box for α-helix and black arrow for β-strand). Bold letters indicate amino acid residues conserved between the two proteins. The amino acid residues shown in red represent the mutated residues in the present study. The amino acid residues shown in blue are the truncated residues 214–228 in Tk-RNase HII-213. The truncation sites are indicated by green triangles. (B) Stereodiagram of the structures of Tk-RNase HII-213 (blue), Mj-RNase HII (red), and Ec-RNase HI (yellow). Root mean square displacements of the superimposition on the Tk-RNase HII structure are 1.2 Å for the 190 Cα atoms of Mj-RNase HII, and 2.0 Å for the 47 Cα atoms consisting of the core β-sheet of Ec-RNase HI. Active-site residues are shown by white stick models. The "basic protrusion" of Ec-RNase HI is indicated by a yellow arrow. (C) A close-up view of the active-site of the three RNases H. The view direction is the same as in the panel B and in Figure 1 ▶.
Figure 2B ▶ shows structural superimposition of Mj-RNase HII (Lai et al. 2000) and Ec-RNase HI (Katayanagi et al. 1992) onto the Tk-RNase HII-213 structure. The main chain structures of the two RNases HII are similar, except for the portions of sequence gaps and the α9 helix at the C-terminal domain. The comparison of the N-terminal domains of the two RNases HII with Ec-RNase HI indicates that only the core β-sheets are well superimposed to each other. In particular, the so-called "basic protrusion" in Ec-RNase HI is absent in both Type 2 enzymes. The region is important for the Type 1 enzyme to bind DNA/RNA hybrid (Katayanagi et al. 1992). Instead, two helices (α1 and α2), which are unique to the two Type 2 RNases, are added on the opposite side of the basic protrusion with respect to the core β-strand (Fig. 2B ▶). This region contains significantly conserved amino acid sequences among known Type 2 enzymes (Haruki et al. 1998). Like the basic protrusion in Ec-RNase HI, this protrusion in the Type 2 enzymes may also participate in binding the substrate, because the conserved region is distinct from the active-site discussed below.
Lai and coworkers (2000) reported that Mj-RNase HII forms a dimer in the crystal. In contrast, the Tk-RNase HII structure is obviously monomeric, even in the crystal. In agreement with this, both analyses by size exclusion chromatography and dynamic light scattering indicated that the Tk-RNase HII is present as a monomer in solution (data not shown). Thus, we believe that dimer formation is not essential, at least, for the RNase H activity.
Active-site
Lai and coworkers (2000) proposed possible active-site residues of Mj-RNase HII by comparing its structure with those of E. coli RNase HI (Ec-RNase HI) (Katayanagi et al. 1992) and retroviral integrase (Bujacz et al. 1995). The sequence alignment between Mj-RNase HII and Tk-RNase HII indicates perfect conservation of the proposed residues (Fig. 2A ▶), suggesting that the catalytic mechanisms of these two enzymes are the same. Actually, their putative active-site residues are well superimposed onto each other with a root-mean-square displacement of 0.67 Å, including the side chain atoms (Fig. 2C ▶). To obtain more convincing experimental evidence for the catalytic residues, we constructed five Tk-RNase HII mutants, in which the putative catalytic residues, Asp7, Glu8, Asp105, His132, and Asp135, were individually replaced by Ala.
The far- and near-UV CD (circular dichroism) spectra of the five mutants were nearly identical to those of the wild-type protein (data not shown), suggesting that the conformation of the protein was not changed markedly by these mutations. Table 1 summarizes the kinetic parameters of the five mutants for the hydrolysis of M13 DNA/RNA hybrid. The D7A and D105A mutants showed no detectable activity. The D135A mutant showed only a trace of the activity. These three residues possess highly conserved carboxyl groups required for polynucleotide transfer reaction (Yang and Steitz 1995; Mizuuchi 1997; Morikawa 1998).
Table 1.
Kinetic parameters of Tk-RNase HII mutants
| Protein | Km (μM) | kcat |
| Wild type | 0.093 | 1.0 |
| D7A | <0.001 | |
| E8A | 1.18 | 0.0015 |
| D105A | <0.001 | |
| H132A | 1.40 | 0.24 |
| D135A | 0.538 | 0.0020 |
| Tk-RNase HII-217 | 0.0974 | 1.15 |
| Tk-RNase HII-213 | 0.190 | 1.60 |
| Tk-RNase HII-207 | 1.55 | 1.41 |
| Tk-RNase HII-203 | 8.37 | 0.18 |
| Tk-RNase HII198 | <0.001 |
Errors, which represent 67% confidence limits, are all at or below ±20% of the values reported.
The E8A mutant also showed only a trace of the activity. This glutamate residue is conserved completely among known Type 2 RNases H (Ohtani et al. 1999a). It is notable that the D135A and E8A mutants showed a greater decrease in kcat than in Km. When the interactions between the mutant proteins (D7A, E8A, D105A, and D135A) and DNA/RNA hybrid were examined using the BIAcore (Pharmacia), their sensorgrams were nearly identical to that of the wild-type protein (data not shown). These results suggest that Asp7, Glu8, Asp105, and Asp135 are involved in the catalytic reaction, rather than the substrate binding.
Kashiwagi and coworkers (1996) proposed a catalytic mechanism of Ec-RNase HI, in which His124 acts as a general base with the aid of Asp134. Assuming the participation of the histidine residue in the catalytic reaction of the Type 2 RNase H, as found in the catalytic reaction of the Type 1 RNase H, His132 is the only possible candidate for the same role. However, the H132A mutant retained substantial activity, although it was lower than that of the wild-type protein (Table 1), indicating that His132 is not essential for the activity. Consistent with this, His132 is conserved only in limited organisms (Ohtani et al. 1999a). As shown in Figure 3 ▶, the side chain of His132 forms hydrogen bonds with the main chain atoms of the loop between the β4 strand and the α5 helix, which contains the catalytically important Asp105. Therefore, it is likely that the decrease in the activity shown by the His132 → Ala protein is attributable to a change of the active-site conformation.
Fig. 3.

A representative 2Fo-Fc electron density map after refinement. The model of a protein portion around His132 is fitted into density contoured at the 1.0 σ level. The imidazole group can assume two conformations, both of which form hydrogen bond(s) with the loop adjacent to the catalytic residue Asp105.
The lack of the catalytic histidine residue in Type 2 RNase H does not necessarily indicate that the catalytic mechanism of Type 2 RNase H differs from that of Type 1 RNase H. The Type 1 and Type 2 enzymes share a common main chain fold (Fig. 2B ▶), and the geometries of the catalytically important acidic residues are well conserved in these two enzymes (Fig. 2C ▶). This highly conserved configuration suggests that the Type 1 and Type 2 RNases H use a similar catalytic mechanism. Kashiwagi and coworkers (1996) proposed that His124 and Asp134 in Ec-RNase HI jointly participate in the activation of a water molecule attacking an RNA phosphate group. Notably, in the superimposition of the RNases H shown in Figure 2C ▶, Asp135 of Tk-RNase HII is located between His124 and Asp134 of Ec-RNase HI. Furthermore, when the structure of Tk-RNase HII is superimposed onto that of the D70N mutant of Ec-RNase HI (Katayanagi et al. 1993), the carboxyl group of Asp135 in Tk-RNase HII occupies a position equivalent to the His124 side chain in Ec-RNase HI (not shown). Hence, we surmise that the catalytic roles of His124-Asp134 in Ec-RNase HI could be performed by the single, invariant Asp135 alone in Tk-RNase HII. This hypothesis is consistent with the observations that Ec-RNase HI can proceed the hydrolytic reaction without His124 (Oda et al. 1993).
It has been proposed that Ser159 of Mj-RNase HII (Ser145 in Tk-RNase HII) is involved in the catalytic reaction, because it is located close to the active-site residues and forms a hydrogen bond with Asp149 (Asp135 in Tk-RNase HII) (Lai et al. 2000). However, this serine residue is not fully conserved in the Type 2 RNase H sequences. For example, Bacillus subtilis RNase HII, which has Ala at the corresponding position, has been shown to exhibit the RNase H activity (Ohtani et al. 1999b). These results strongly suggest that the serine residue is not involved in the catalytic reaction of Type 2 RNase H.
The endonulease activity of Tk-RNase HII requires a divalent metal cation, such as Mg2 +, Mn2 +, or Co2 + (Haruki et al. 1998). To identify metal binding sites, these metal ions were soaked into the crystal. However, no significant electron density was observed around the active-site in difference Fourier maps. Thus, the affinity of the metals with Tk-RNase HII appears to be low in the absence of DNA/RNA hybrids.
Substrate binding
Limited proteolytic analyses indicated that the segment 199–217 is important for the RNase H activity. To investigate the functional role of this segment, the enzymatic properties of the five truncated proteins were analyzed. The far-UV CD spectra of these truncated proteins were nearly identical to that of the wild-type protein (data not shown), suggesting that the protein conformation was not markedly changed by the C-terminal truncations. The kinetic parameters of the truncated proteins are compared with those of the wild-type protein in Table 1. The kinetic parameters of Tk-RNase HII-217 were almost identical to those of the wild-type protein. The additional truncation of the four residues did not seriously affect the Km and kcat values. However, further truncations resulted in a dramatic increase in the Km value. They affected the kcat value as well, but much less seriously. As a result, the Km values of Tk-RNase HII-207 and Tk-RNase HII-203 were increased by 17- and 90-fold, respectively, as compared to that of the wild-type protein. Thus, the deletion of residues 199–213, which form the α9 helix (Fig. 1 ▶), seriously affected the Km value, rather than the kcat value. The interactions between the truncated proteins and the substrate were analyzed using surface plasmon resonance (Fig. 4 ▶). Tk-RNase HII-213 and Tk-RNase HII-217 with the intact α9 helix retained almost full substrate binding affinity. In contrast, this affinity decreased significantly as the length of the α9 helix was reduced by the truncation. Consequently Tk-RNase HII-198, which completely lost the α9 helix, did not bind to the substrate at all. Tk-RNase HII-203 apparently did not bind to the substrate, although it retained RNase H activity. The interaction between this protein and the substrate may be too weak to be detected by the BIAcore analysis. Taken together, these results suggest strongly that the α9 helix plays an important role in substrate binding. Because this helix contains many basic residues (Fig. 2A ▶), it is likely to contact the negatively charged DNA/RNA hybrid by electrostatic force.
Fig. 4.

Surface plasmon resonance analysis of the proteins (100 nM) as reported by Haruki and coworkers (1997). Black, blue, green, orange, red, and cyan lines are sensorgrams for the wild-type protein, Tk-RNase HII-217, Tk-RNase HII-213, Tk-RNase HII-207, Tk-RNase HII-203, and Tk-RNase HII-198, respectively.
Nakamura and coworkers (1991) built a model of the Ec-RNase HI-DNA/RNA hybrid complex based on NMR titration experiments and mutational studies. As the architectures of the major core regions are well conserved between Ec-RNase HI and Tk-RNase HII, a docking examination could address the question of whether the α9 helix indeed participates in the substrate binding. The superimposition of Tk-RNase HII-213 structure onto the Ec-RNase HI-DNA/RNA hybrid allowed the construction of a complex model without any steric hindrance against the substrate (Fig. 5A ▶). The overall configuration of the DNA/RNA hybrid with respect to the N-terminal domain agrees with the structure of a DNA duplex bound to HIV-1 reverse transcriptase (PDB accession code: 1C9R; Ding et al. 1998). However, it appears that the basic helical region is too far from the substrate to make direct contact. We presume that this region would move to make electrostatic interactions with the substrate. For instance, the rotation of the C-terminal domain by ∼70° toward the substrate will allow the α9 helix to contact the phosphate backbone of the DNA/RNA hybrid, as shown in Figure 5B ▶.
Fig. 5.

(A) A hypothetical docking model of the DNA/RNA hybrid on Tk-RNase HII-213. This model was generated by superposition of the obtained crystal structure onto the Ec-RNase HI-DNA/RNA model proposed by Nakamura and coworkers (1991). Yellow and green ribbons indicate RNA and DNA strands, respectively. Blue and red surfaces represent the segment 199–213 and the active-site residues, respectively. (B) A putative substrate recognition by Tk-RNase HII. The C-terminal domain is rotated by ∼70° so that the basic α9 helical segment can make contact with phosphate backbones of the DNA/RNA hybrid duplex.
Materials and methods
Preparation of the protein
The construction of an E. coli overexpression strain for Tk-RNase HII, and its overproduction and purification were performed as reported by Haruki and coworkers (1998).
Briefly, the E. coli cells that overproduced the protein were harvested and suspended in 10mM Tris-HCl at pH7.5 containing 1mM EDTA. After sonication and then incubation at 90°C, nucleic acid was removed by polyethyleneimine treatment, followed by ammonium sulfate precipitation (70% sat.). Finally, the protein was purified to homogeneity by chromatography on HiTrap heparin and MonoQ columns (Amersham Pharmacia Biotech). The purity was confirmed by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The concentrations of the wild-type, mutant, and truncated proteins were estimated by assuming A280nm of 0.63 for 1 mg/ml protein.
The five C-terminal truncated proteins were constructed by introducing a stop codon (TAA) at the corresponding position. Overproduction and purification of these truncated proteins were performed as reported for the wild-type protein (Haruki et al. 1998).
X-ray crystallography
Tk-RNase HII-213 was crystallized by the hanging drop vapor diffusion method with 15%(w/v) PEG6000 and 200 mM MES at pH 6.5. Two sets of X-ray diffraction data were collected for the native crystal (Table 2). The initial phases were estimated by the MIR method using the first data set and three kinds of heavy atom derivatives (Table 2). The density modification (Cowtan 1994) extended the resolution to 3.5 Å and effectively improved the electron density map. After 80 % of the amino acid residues were assigned, the structure factor data were swapped for the second data set, because of the higher resolution limit. The CNS refinement (Brünger et al. 1998) with manual model correction reduced the R-factor and the free R to 0.233 and 0.278, respectively, with the 15–2.0 Å resolution data. The refinement and the final model statistics are summarized in Table 3. The atomic coordinates have been deposited to the Protein Data Bank (accession code: 1IO2).
Table 2.
X-ray data collection and MIR phasing statistics
| Native | Derivative | ||||
| Data collection | Data 1 | Data 2 | K2Pt(NO3)4 | SmCl3 | K2OsO4 |
| X-ray source | CuKαa | synchrotronb | CuKαa | CuKαa | CuKαa |
| Resolution (Å) | 50–2.4 | 50–2.0 | 50–2.6 | 50–2.6 | 50–2.4 |
| Observed reflections | 73341 | 52662 | 31026 | 25937 | 41799 |
| Unique reflections | 10457 | 15205 | 8080 | 8040 | 9896 |
| Rmergec (%) | 5.7 (14.8d) | 5.4 (25.4e) | 9.5 (31.5f) | 11.6 (36.4f) | 6.5 (26.8d) |
| Completeness (%) | 99.2 (91.9d) | 86.6 (73.7e) | 98.4 (97.7f) | 98.5 (99.9f) | 96.9 (97.8d) |
| Ilσ(I) | 25.8 (12.3d) | 17.8 (4.58e) | 9.13 (3.55f) | 7.00 (2.89f) | 12.4 (4.27d) |
| MIR phasingg | |||||
| Resolution (Å) | 15–5.0 | 15–5.0 | 15–5.0 | 15–5.0 | |
| Phasing power | 1.91 | 1.45 | 1.49 | ||
| Rcullis | 0.51 | 0.56 | 0.58 | ||
| Figure of merit | 0.822 | ||||
a With DIP100 image plate detector (MacScience).
b At the BL6A2 beamline (λ = 1.000 [Å]) of the Photon Factory with the Sakabe Weissenberg camera (Sakabe 1991).
c By the HKL suite (Otwinowski and Minor 1997).
d 2.49–2.40 Å resolution.
e 2.07–2.00 Å resolution.
f 2.69–2.60 Å resolution.
g By the program MLPHARE (Otwinowski 1991) in the CCP4 suite (Collaborative Computational Project, Number 4, 1994).
Table 3.
Refinement and model statistics
| Resolution (Å) | 15–2.0 |
| Rwork | 0.223 (0.218b) |
| Rfreea | 0.274 (0.275b) |
| Number of atoms | |
| Protein | 1652 |
| Solvent | 216 |
| Average B-factor (Åb) | |
| Protein | 20.4 |
| Solvent | 33.3 |
| Rms deviation from ideal | |
| Bone length (Å) | 0.005 |
| Bond angle (°) | 1.16 |
| Ramachandran plotc (%) | |
| Most favored | 95.7 |
| Additionally allowed | 4.3 |
| Generously allowed | 0.0 |
| Disallowed | 0.0 |
a 5% of reflections not used throughout the structure refinement.
b Statistics for 2.03–2.00 Å resolution.
c Laskowski (1993).
Mutational analysis
Site-directed mutagenesis was performed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Oligonucleotides for the mutageneses were synthesized by Sawady Technology Co. Ltd. (Japan). They were designed to alter the codons for Asp7, Glu8, Asp105, His132, and Asp135 to those for Ala. The RNase H assay was performed at 30°C and pH 8.0 using DNA/RNA hybrid as a substrate, as described by Haruki and coworkers (1998). For the kinetic analyses, the substrate concentration spanned the Km values. The hydrolysis of the substrate with the enzyme followed Michaelis-Menten kinetics, and the kinetic parameters were determined from a Lineweaver-Burk plot. The CD spectra were measured in 10 mM Tris-HCl at pH 8.0 at 25°C on a J-725 automatic spectropolarimeter (Japan Spectroscopic Co. Ltd.), as reported previously (Hirano et al. 1998). To measure the substrate binding at pH 7.5 in the absence of a divalent cation, BIAcore surface plasmon resonance analysis was performed according to the manufacturer's recommendations (Pharmacia). The immobilization of the 36-bp DNA/RNA hybrid onto the sensor chips was performed as reported previously (Haruki et al. 1997)
Acknowledgments
We thank Mr. Kochi for technical assistance with biochemical experiments.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
RNase, ribonuclease
DNA, deoxyribonucleic acid
RNA, ribonucleic acid
MIR, multiple isomorphous replacement
PEG, polyethylene glycol
MES, 2-(N-morpholino)ethanesulfonic acid
MALDI, matrix-assisted laser desorption ionization
TOF, time-of-flight
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.48001.
References
- Ariyoshi, M., Vassylyev, D.G., Iwasaki, H., Nakamura, H., Shinagawa, H., and Morikawa, K. 1994. Atomic structure of the RuvC resolvase: A Holliday junction-specific endonuclease from E. coli. Cell 82 209–220. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system (CNS): A new software suite for macromolecular structure determination. Acta Cryst. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Bujacz, G., Jaskolski, M., Alexandratos, J., and Wlodawer, A. 1995. High-resolution structure of the catalytic domain of avian sarcoma virus integrase. J. Mol. Biol. 253 333–346. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Cryst. D50 760–763. [DOI] [PubMed] [Google Scholar]
- Cowtan, K.D. 1994. "dm": An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsl. Protein Crystallogr. 31 34– 38. [Google Scholar]
- Davies, J.F.D., Hostomska, Z., Hostomsky, Z., Jordan, S.R., and Matthews, D.A. 1991. Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 252 88–95. [DOI] [PubMed] [Google Scholar]
- Ding, J., Das, K., Hsiou, Y., Sarafianos, S.G., Clark, A.D. Jr., Jacobo-Molina, A., Tantillo, C., Hughes, S.H., and Arnold, E. 1998. Structure and functional implications of the polymerase active-site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol. 284 1095–1111. [DOI] [PubMed] [Google Scholar]
- Dyda, F., Hickman, A.B., Jenkins, T.M., Engelman, A., Craigie, R., and Davis, D.R. 1994. Crystal structure of the catalytic domain of HIV-1 integrase: Similarity to other polynucleotidyl transferase. Science 266 1981–1986. [DOI] [PubMed] [Google Scholar]
- Haruki, M., Noguchi, E., Kanaya, S., and Crouch, R.J. 1997. Kinetic and stoichiometric analysis for the binding of Escherichia coli Ribonuclease HI to RNA-DNA hybrids using surface plasmon resonance. J. Biol. Chem. 272 22015–22022. [DOI] [PubMed] [Google Scholar]
- Haruki, M., Hayashi, K., Kochi, T., Muroya, A., Koga, Y., Morikawa, M., Imanaka, T., and Kanaya, S. 1998. Gene cloning and characterization of recombinant RNase HII from a hyperthermophilic archaeon. J. Bacteriol. 180 6207–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausen, P. and Stein, H. 1970. Ribonuclease H. An enzyme degrading the RNA moiety of DNA-RNA hybrids. Eur. J. Biochem. 14 278–283. [DOI] [PubMed] [Google Scholar]
- Hirano, N., Haruki, M., Morikawa, M., and Kanaya, S. 1998. Stabilization of ribonuclease HI from Thermus thermophilus HB8 by the spontaneous formation of an intramolecular disulfide bond. Biochemistry 37 12640–12648. [DOI] [PubMed] [Google Scholar]
- Ishikawa, K., Okumura, M., Katayanagi, K., Kimura, S., Kanaya, S., Nakamura, H., and Morikawa, K. 1993. Crystal structure of ribonuclease H from Thermus thermophilus HB8 at 2.8 Å resolution. J. Mol. Biol. 230 529–542. [DOI] [PubMed] [Google Scholar]
- Kanaya, S. 1998. Enzymatic activity and protein stability of E. coli ribonuclease HI. In Ribonucleases H (eds. R.J. Crouch and J.J. Toulmé), pp. 1–38. Les Editions INSERM, Paris.
- Kashiwagi, T., Jeanteur, D., Haruki, M., Katayanagi, K., Kanaya, S., and Morikawa, K. 1996. Proposal for new catalytic roles for two invariant residues in Escherichia coli ribonuclease HI. Protein Eng. 9 857–867. [DOI] [PubMed] [Google Scholar]
- Katayanagi, K., Miyagawa, M., Ishikawa, M., Matsushima, M., Kanaya, S., Ikehara, M., Matsuzaki, T., and Morikawa, K. 1990. Three-dimensional structure of ribonuclease H from E. coli. Nature 347 306–309. [DOI] [PubMed] [Google Scholar]
- Katayanagi, K., Miyagawa, M., Matsushima, M., Ishikawa, M., Kanaya, S., Nakamura, H., Ikehara, M., Matsuzaki, T., and Morikawa, K. 1992. Structural details of ribonuclease H from Escherichia coli as refined to an atomic resolution. J. Mol. Biol. 223 1029–1052. [DOI] [PubMed] [Google Scholar]
- Katayanagi, K., Ishikawa, M., Okumura, M., Ariyoshi, M., Kanaya, S., Kawano, Y., Suzuki, M., Tanaka, I., and Morikawa, K. 1993. Crystal structures of ribonuclease HI active-site mutants from Escherichia coli. J. Biol. Chem. 268 22092–22099. [PubMed] [Google Scholar]
- Kogoma, T. and Foster, P.L. 1998. Physiological functions of Escherichia coli RNase HI. In Ribonucleases H (eds. R.J. Crouch and J.J. Toulmé), pp. 39–66. Les Editions INSERM, Paris.
- Lai, L., Yokota, H., Hung, L.W., Kim, R., and Kim, S.H. 2000. Crystal structure of archaeal RNase HII: A homologue of human major RNase H. Structure 8 897–904. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Mizuuchi, K. 1997. Polynucleotidyl transfer reaction in site-specific DNA recombination. Genes Cells 2 1–12. [DOI] [PubMed] [Google Scholar]
- Morikawa, K. 1998. Crystallographic studies of proteins involved in recombinatorial repair and excision repair. In Nucleic Acids and Molecular Biology (eds. F. Eckstein and D.M.J. Lilley), pp. 275–299. Springer-Verlag. Berlin, Germany.
- Morikawa, K. and Katayanagi, K. 1998. Crystal structure of RNase H from prokaryotes. In Ribonucleases H (eds. R.J. Crouch and J.J. Toulmé), pp. 181–193. Les Editions INSERM, Paris.
- Nakamura, H., Oda, Y., Iwai, S., Inoue, H., Ohtsuka, E., Kanaya, S., Kimura, S., Katsuda, C., Katayanagi, K., Morikawa, K., et al. 1991. How does RNase H recognize a DNA-RNA hybrid. Proc. Natl. Acad. Sci. USA 88 11535–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda, Y., Yoshida, M., and Kanaya, S. 1993. Role of histidine 124 in the catalytic function of ribonuclease HI from Escherichia coli. J. Biol. Chem. 268 88–92. [PubMed] [Google Scholar]
- Ohtani, N., Haruki, M., Morikawa, M., and Kanaya, S. 1999a. Molecular diversities of RNase H. J. Biosci. Bioeng. 88 12–19. [DOI] [PubMed] [Google Scholar]
- Ohtani, N., Haruki, M., Morikawa, M., Crouch, R.J., Itaya, M., and Kanaya, S. 1999b. Identification of the genes encoding Mn2 +-dependent RNase HII and Mg 2+-dependent RNase HIII from Bacillus subtilis: Classification of RNase H into three families. Biochemistry 38 605–618. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. 1991. Maximum likelihood refinement of heavy atom parameters. In Proceedings of the CCP4 study weekend: "Isomorphous replacement and anomalous scattering" (eds. W. Wolf, P.R. Evans, and A.G.W. Leslie), pp. 80–86. SERC Daresbury Laboratory, Daresbury, UK.
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Rice, P. and Mizuuchi, K. 1995. Structure of bacteriophage Mu transposase core: A common structural motif for DNA transposition and retroviral integration. Cell 82 209–220. [DOI] [PubMed] [Google Scholar]
- Sakabe, N. 1991. X-ray diffraction data collection system for modern protein crystallography with a Weissenberg camera and an imaging plate using synchrotron radiation. Nucl. Inst. Meth. Phys. Res. A303 448–463. [Google Scholar]
- Yang, W. and Steitz, T.A. 1995. Recombining the structure of HIV integrase, RuvC and RNase H. Structure 3 131–134. [DOI] [PubMed] [Google Scholar]
- Yang, W., Hendrickson, W.A., Crouch, R.J., and Satow, Y. 1990. Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenomethionyl protein. Science 249 1398–1405. [DOI] [PubMed] [Google Scholar]