

Abstract
A high resolution NMR structure of hen lysozyme has been determined using 209 residual 1H–15N dipolar coupling restraints from measurements made in two different dilute liquid crystalline phases (bicelles) in conjunction with a data set of 1632 NOE distance restraints, 110 torsion angle restraints, and 60 hydrogen bond restraints. The ensemble of 50 low-energy calculated structures has an average backbone RMSD of 0.50±0.13Å to the mean structure and of 1.49±0.10Å to the crystal structure of hen lysozyme. To assess the importance of the dipolar coupling data in the structure determination, the final structures are compared with an ensemble calculated using an identical protocol but excluding the dipolar coupling restraints. The comparison shows that structures calculated with the dipolar coupling data are more similar to the crystal structure than those calculated without, and have better stereochemical quality. The structures also show improved quality factors when compared with additional dipolar coupling data that were not included in the structure calculations, with orientation-dependent 15N chemical shift changes measured in the bicelle solutions, and with T1/T2 values obtained from 15N relaxation measurements. Analysis of the ensemble of NMR structures and comparisons with crystal structures, 15N relaxation data, and molecular dynamics simulations of hen lysozyme provides a detailed description of the solution structure of this protein and insights into its dynamical behavior.
Keywords: Bicelles, biomolecular NMR, lysozyme, protein structure, Q factor, residual dipolar couplings, simulated annealing
Hen lysozyme is one of the most studied and best characterized globular proteins. It was the first enzyme to have its structure determined by X-ray diffraction (Blake et al. 1965), and since then hen lysozyme has been used extensively as a system in which to understand the underlying principles of protein structure, function, dynamics, and folding through studies of both an experimental and theoretical nature (Jollès and Jollès 1984; Johnson et al. 1988; Dobson et al. 1994; Buck et al. 1995; Smith et al. 1995; Krebs et al. 2000). Hen lysozyme gives good quality NMR spectra (Redfield and Dobson 1988), but until now it has not been possible to obtain a high resolution structure of the protein in solution by NMR techniques. Such a structure is, however, important. When combined with insight from experiment and theory, it provides a basis for understanding in detail the properties of the protein that depend on its dynamical behavior in solution.
The previously experienced challenges with determining an NMR structure of lysozyme at high resolution reflect to a large extent the characteristics of the lysozyme structure, which has two structural domains: the α-domain (residues 1–35, 85–129; four α-helices and a short 310-helix) and the β-domain (residues 36–84; a triple-stranded antiparallel β-sheet, a long loop, and a 310-helix). The α-domain contains a core of hydrophobic side-chains that are packed closely together (the hydrophobic box); the structure of this domain is relatively well defined in an ensemble of structures of lysozyme calculated using NMR data from homonuclear studies reported previously (Smith et al. 1993; mean Cα RMSD for the α-domain of 1.2±0.2 Å with respect to the average structure). In contrast, in the β-domain of the protein there is no similar hydrophobic core; instead, hydrogen bonds and a number of small hydrophobic clusters appear to be responsible for defining the tertiary fold, and there is also a long exposed loop region. Few long-range NOEs could be identified in the 1H spectra for residues in the β-domain of lysozyme; excluding those that define β-strands or bridges, only 68 long-range NOEs (i,i+5 or greater) for residues in the β-domain were available for the structure determination. This presumably reflects the absence of a large hydrophobic core. Consequently, in the initial NMR structures, each of the different regions of the β-domain are well defined locally but their relative orientations are not (Smith et al. 1993; mean Cα RMSD of 2.2±0.4 Å for the β-domain with respect to the average structure).
The problem of defining the structure of the β-domain in the initial NMR structures is indicated clearly in Figure 1a ▶, in which backbone RMSD values of greater than 3Å from the mean structure can be seen for residues in the regions Asn 46-Gly 49, Arg 68-Pro 70 and Ser 81-Leu 83. Experimental 15N relaxation measurements for hen lysozyme (Buck et al. 1995) show that the main-chain amide groups of most residues in the protein undergo only small amplitude librational motions on a fast timescale with order parameters greater than 0.8 (calculated using a N-H bond length of 1.02Å; Case [1999] has shown that with this bond length the maximum order parameter for a model peptide at very low temperature would be 0.86). Four of the residues in the three regions of greatest disorder in the NMR ensemble have slightly lower order parameters in the range 0.7 to 0.8 (Thr 47, Asp 48, Arg 68, Thr 69). However, the extent of disorder observed in the NMR structures for these regions is much larger than would be expected on the basis of their mobility within native lysozyme. Moreover, the residues Ser 81-Leu 83 are in a 310-helix in the X-ray structure of the protein but are not well defined in the initial NMR structure because of a lack of restraints (Smith et al. 1993).
Fig. 1.

Comparison of (a) the average backbone RMSD values (in Å) from the mean structure and (b) the backbone RMSD values (in Å) between the mean structure and the X-ray structure (pdb code 193L) for the refined structures of hen lysozyme reported here and for those reported previously (Smith et al. 1993). In each case, the RMSD values are shown as a function of residue number, (filled circles) the ensemble of 50 refined structures in set 1 reported here, (open circles) the set of 16 structures reported previously (Smith et al. 1993). (c) shows the temperature factors (in Å2) of main-chain N atoms in the X-ray structure of Vaney et al. (1996) with pdb code 193L.
There has been much interest in the use of dipolar coupling data in NMR protein structure determinations (Tjandra and Bax 1997; Clore and Gronenborn 1998; Prestegard 1998). These data establish the orientation of internuclear vectors with respect to an alignment tensor axis frame in the molecule and so complement the short-range NOE and torsion angle restraints. Significant improvements to the accuracy of NMR structures on inclusion of dipolar coupling restraints have been reported for a number of systems, including ubiquitin (Bax and Tjandra 1997), a complex of the transcription factor GATA-1 with DNA (Tjandra et al. 1997), GAIP (de Alba et al. 1999), S4Δ41 (Markus et al. 1999), and a complex of DNA with three zinc fingers (Tsui et al. 2000). In this paper we describe use of such data for hen lysozyme and show that this results in a substantially improved structure.
Results and Discussion
Experimental restraints and structure calculations
1H and 15N chemical shift assignments for native hen lysozyme have been reported previously (Redfield and Dobson 1988; Buck et al. 1995). 13C resonance assignments were made using HNCA, HNCO, HCCH-TOCSY, and (H)CCH-TOCSY spectra (Fesik et al. 1990; Kay et al. 1993; Sattler et al. 1995) recorded using a double-labeled (13C,15N) lysozyme sample (Fig. 2 ▶). These assignments have been deposited in BioMagRes Bank (BMRB accession number 4831). NOEs were identified in 15N NOESY-HMQC and 13C NOESY-HMQC spectra (Driscoll et al. 1990; Ikura et al. 1990). The lysozyme NMR structures of Smith et al. (1993) were used to solve any ambiguities in the NOE assignments resulting from chemical shift degeneracy. NOE intensities were estimated by measuring peak heights and were used to assign interproton distance ranges to the NOE restraints as described in the Materials and Methods section. The final NOE data set consists of 1632 restraints (Table 1), 1096 of these coming from the previous homonuclear NMR studies (Smith et al. 1993).
Fig. 2.

Hα-Cα region of the 600 MHz 1H-13C HSQC spectrum of hen lysozyme at pH 3.8 and 35°C.
Table 1.
Summary of experimental data used in the structure calculations
| Number of restraints | |
| NOE distance restraintsa | |
| Total | 1632 |
| Intraresidue | 560 |
| Sequential | 440 |
| Short range (l <│i − j│ <5) | 279 |
| Long range (│i − j│ ≥5) | 353 |
| Hydrogen bond restraints | 60 |
| Dihedral angle restraints φ | 51 |
| χ1 | 59 |
| Residual dipolar coupling restraints | |
| 5% DMPC:DHPC (2.9:1.0) | 107 |
| 7.5% DMPC:DHPC:CTAB (2.9:1.0:0.1) | 102 |
a On the basis of the heteronuclear NMR data 45 of the homonuclear NOE's used in the structure calculations reported in 1993 were discarded; another 17 of the homonuclear NOE's were not included in the structure calculations either because they did not provide structurally significant restraints or because they were replaced by restraints involving stereospecifically assigned β protons.
3J(HN,Hα) and 3J(Hα,Hβ) coupling constant data measured using homonuclear NMR methods for hen lysozyme have been reported (Smith et al. 1991; Bartik and Redfield 1993). Additional coupling constant values that could not be obtained previously because of low signal intensity or resonance overlap were determined using an HMQCJ experiment (Kay and Bax 1990) on a 15N-labeled lysozyme sample for 3J(HN,Hα) coupling constants and a soft HCCH-E.COSY experiment (Eggenberger et al. 1992) for 3J(Hα,Hβ) side-chain coupling constants. 3J(Hβ,C′) coupling constants were also measured for 45 residues in hen lysozyme using a soft HCCH-COSY experiment (Eggenberger et al. 1992). The coupling constants were converted into dihedral angle restraints as described in the Materials and Methods section, 51 φ and 59 χ1 torsion angle restraints being obtained.
1H-15N residual dipolar couplings were measured for lysozyme in two different bicelle solutions, one containing 5% DMPC:DHPC (2.9:1.0) and the other 7.5% DMPC: DHPC:CTAB (2.9: 1.0: 0.1). Details of sample preparation are given in the Materials and Methods section. Residual dipolar couplings were measured for 107 residues in the 5% DMPC:DHPC bicelles and for 102 residues in the 7.5% DMPC:DHPC:CTAB bicelles. Measurements were not possible for 19 and 24 residues in the 5% and 7.5% bicelle solutions, respectively, either as a result of peak overlap or the absence of peaks at pH 6.5 resulting from exchange with the solvent.
A region of the NMR spectra acquired at 34.5°C for an isotropic solution, and for bicelle solutions with 5% DMPC:DHPC and 7.5% DMPC:DHPC:CTAB, is shown in Figure 3 ▶. It is interesting to note from these spectra, and from the histograms shown in Figure 4 ▶, that the spread of the residual dipolar couplings measured in 5% DMPC: DHPC is larger than that measured in 7.5% DMPC: DHPC:CTAB. An increase in bicelle concentration from 5% to 7.5% should lead to a 50% increase in the magnitude of dipolar couplings if the alignment arises from only steric factors (Bax and Tjandra 1997; Zweckstetter and Bax 2000). The observed decrease in dipolar couplings shows the influence of electrostatic effects on the partial alignment of proteins in bicelle solutions. The results for the 5% DMPC:DHPC solution are consistent with the observation of Losonczi and Prestegard (1998); these authors suggest that the DMPC:DHPC bicelles are not neutral but have a slight negative charge arising from some hydrolysis of the DMPC. Lysozyme is a positively charged protein at pH 6.5 and, therefore, an electrostatic attraction between lysozyme and the bicelle surface will contribute to the orientation of the protein and lead to larger than expected dipolar couplings. The addition of positively charged cetyltrimethylammonium bromide (CTAB) to the bicelles will remove the electrostatic attraction and lead to alignment based on steric factors alone (Zweckstetter and Bax 2000). It is clear from an inspection of Figure 3 ▶ that the orientation of the alignment tensor in the two bicelle solutions differs. The measured residual dipolar coupling for Arg 125 has a different sign in the two solutions. For Ala 82 a coupling of −0.5 Hz is observed in the 5% bicelles and a value of 10.3 Hz in the less strongly aligned 7.5% CTAB containing bicelles. The use of two sets of dipolar couplings, with different alignment tensors, in structure refinement is useful for resolving ambiguities that can arise if only a single set of data is used (Ramirez and Bax 1998). In the structure calculations described below, the orientation of the principle component of the alignment tensors for the two bicelle solutions are found to differ by 9.2±0.3°.
Fig. 3.

Part of the 750 MHz spectrum of hen lysozyme obtained in (a) isotropic solution, (b) 5% DMPC:DHPC (2.9:1.0) bicelles, and (c) 7.5% DMPC:DHPC:CTAB (2.9:1.0:0.1) bicelles. The numbers shown are the measured splittings and, in brackets, the residual dipolar couplings.
Fig. 4.
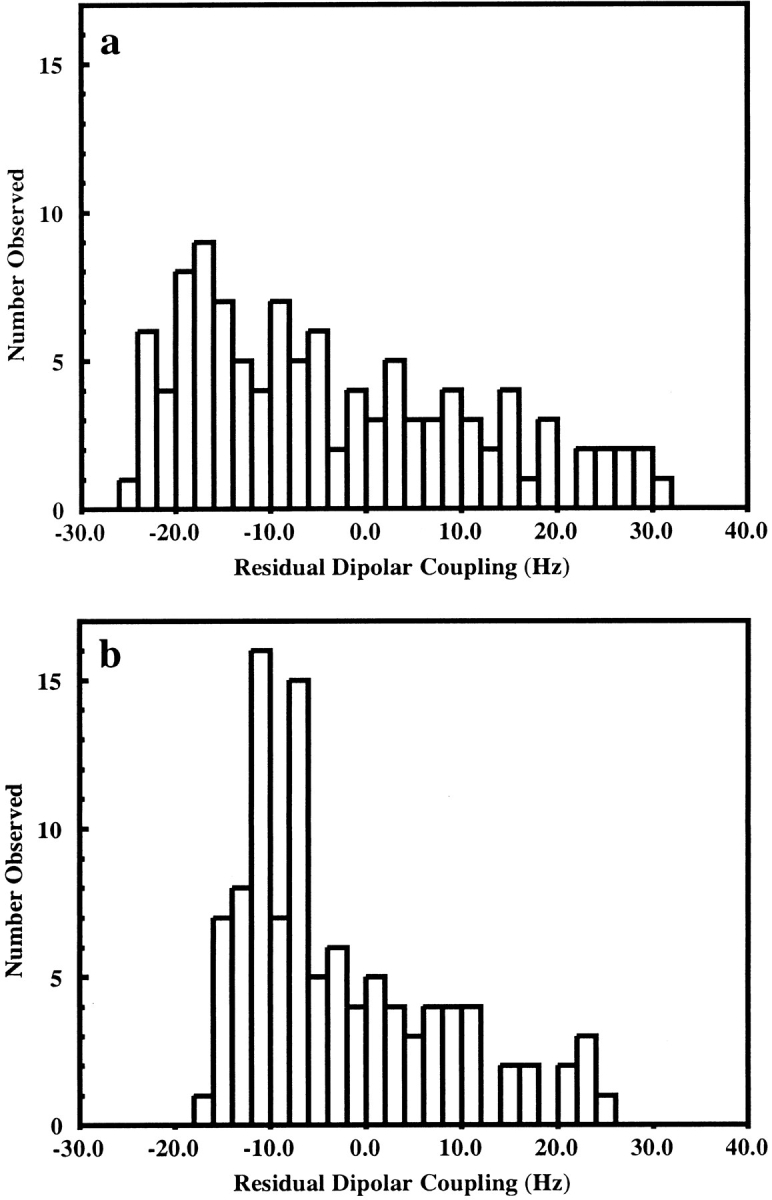
Histograms showing the distribution of residual dipolar couplings measured for lysozyme in (a) 5% DMPC:DHPC and (b) 7.5% DMPC: DHPC:CTAB.
The refinement of a protein structure using additional residual dipolar coupling restraints and the program XPLOR requires that information about the alignment tensor be specified. Clore et al. (1998a) have shown that the axial and rhombic components of the alignment tensor (Da and Dr) can be estimated from the high and low extreme values and from the most populated value in a histogram showing the distribution of residual dipolar couplings. The distributions of residual dipolar couplings measured for the 5% DMPC:DHPC and 7.5% DMPC:DHPC:CTAB bicelles are plotted in Figure 4 ▶. In both cases it is difficult to determine the most populated value of the dipolar coupling from the histograms and, therefore, the estimates of Da and R (R = Dr/Da) have been determined from the high and low extreme values only; for the 5% DMPC:DHPC solution Da = 15.1 and R = 0.34, whereas for the 7.5% DMPC: DHPC:CTAB solution Da = 12.2 and R = 0.16. The larger rhombicity (R) for 5% DMPC:DHPC bicelles is consistent with a larger electrostatic contribution to the orientation of the protein (Sass et al. 1999). The values of Da and R were further refined using the procedure of Clore et al. (1998b) as described in Materials and Methods.
Using the NOE, hydrogen bond, and torsion angle restraints for hen lysozyme summarized in Table 1, a set of structures for hen lysozyme was calculated with an extended simulated annealing protocol (Nilges et al. 1988; Wiles et al. 1997), and the 15 lowest energy structures were selected. From each of these structures, 20 conformers were calculated using the 209 restraints from the residual dipolar coupling data using the protocol described in the Materials and Methods section. The 50 lowest energy structures from these calculations were then analyzed (structure set 1). The structural statistics for the final ensemble of structures are given in Table 2. The ensemble of structures and the input NMR restraints have been deposited in the Brookhaven Protein Databank (code 1E8L).
Table 2.
Structural statistics for the calculated ensembles of 50 low-energy structures
| With dipolar couplings (Structure set 1) | Without dipolar couplings (Structure set 2) | |
| RMS deviations from ideal covalent geometry | ||
| Bonds (Å) | 0.00333 ± 0.00007 | 0.00252 ± 0.000021 |
| Angles (deg.) | 0.492 ± 0.0069 | 0.347 ± 0.0018 |
| Impropers (deg.) | 0.384 ± 0.0079 | 0.247 ± 0.0037 |
| RMS deviations from experimental restraintsa | ||
| NOE (Å) | 0.0439 ± 0.0009 | 0.0350 ± 0.00025 |
| Dihedrals (deg.) | 0.655 ± 0.045 | 0.412 ± 0.0227 |
| Dipolar (Hz) | 1.159 ± 0.042 | |
| Structural quality and coordinate precision | ||
| Backbone RMSD to mean (Å) residues 1–129 | 0.50 ± 0.13 | 0.60 ± 0.14 |
| Backbone RMSD to X-ray (Å) residues 1–129 | 1.49 ± 0.10 | 1.69 ± 0.12 |
| Backbone RMSD to mean (Å) for the 117 well defined residuesb | 0.43 ± 0.10 | 0.42 ± 0.07 |
| Backbone RMSD to X-ray (Å) for the 117 well defined residuesb | 1.32 ± 0.08 | 1.50 ± 0.08 |
| Percentage of residues in the most favoured regions of the Ramachandran plotc | 74.2 ± 2.0 | 65.9 ± 2.6 |
a For each of the structures in set 1, there are 0 or 1 NOE violations >0.5 Å, 0 or 1 dihedral violations >5°, and 3–8 dipolar violations >3 Hz. None of the structures in set 2 have NOE or dihedral violations >0.5 Å or 5°, respectively.
b The residues excluded are those where for the ensemble of NMR structures in set 1 there is a backbone RMSD to the mean structure >0.8 Å (residues 22, 46–48, 68–71, 102, 128–129) or where the RMSD between the mean NMR structure from set 1 and the X-ray structure is >3 Å (residue 117).
c Calculated using the program PROCHECK (Laskowski et al. 1993).
Characteristics of the NMR structure of hen lysozyme in solution
The mean structure calculated from the ensemble of NMR structures for hen lysozyme is shown in Figure 5A ▶. The quality of the structure is very significantly improved compared with the structure reported previously (mean backbone RMSD to the average structure is 0.50±0.13Å compared with 1.71±0.25Å for the 1993 structures). The RMSD of the NMR structures to the crystal structure of hen lysozyme of Vaney et al. (1996; structure of the tetragonal form at 1.33Å with pdb code 193L; referred to here as the X-ray structure) is also reduced (mean backbone RMSD is 1.49±0.10Å compared with 2.33±0.33Å for the 1993 structures). There is also a substantial improvement in the stereochemical quality; an analysis using PROCHECK (Laskowski et al. 1993) showed that 74.2% of the φ,ψ; torsion angles of residues in the protein lie in the most favored regions of the Ramachandran plot (compared with 54.5% for the 1993 structures).
Fig. 5.

Ribbon diagrams showing the mean structures of hen lysozyme from sets 1 (A) and 2 (B). In each case the radius of the ribbon shows the rms difference from the mean for the Cα atoms across the set of 50 structures. Residues 6–15, 25–36 and 89–100, which form the three main α-helices in the protein (helices A, B and C), are shown in red. Three shorter regions of helix are shown in cyan. These correspond to helix D (residues 111–114) and the two 310-helices (residues 81–84 and 120–123) in the X-ray structure. Residues 41–60, which form the triple stranded antiparallel β-sheet in the X-ray structure, are shown in yellow. The N and C termini are labeled. The figure was generated using MOLMOL (Koradi et al. 1996).
Regions of secondary structure in the NMR structures have been identified according to the criteria of Kabsch and Sander (1983). In agreement with the X-ray structure, three long α-helices are present in all members of the family of NMR structures (A helix: Cys 6-His 15; B helix: Leu 25-Ser 36; C helix: Thr 89-Ser 100). The fourth α-helix (D) in the X-ray structure is present in 41 of the 50 NMR structures, although it is slightly reduced in length (Trp 111-Arg 114 compared with Val 109-Arg 114 in the X-ray structure); a series of turns is formed for this sequence in the other nine NMR structures. A β-bridge involving Val 2 and Asn 39 is present in all of the NMR structures (also in the X-ray structure), but only the first two strands of the triple stranded antiparallel β-sheet present in the X-ray structure are defined in the NMR structures (Thr 43-Arg 45 and Thr 51-Tyr 53; in 14 of the structures the length of these strands is reduced). This irregularity in the β-sheet may result from slight variations in the relative positions of NH and CO groups because of the inclusion of the dipolar coupling restraints in the structure calculations, which we discuss in the next section. Helices are also identified in the NMR structures for Ser 81-Leu 83/84 (310-helix in 30 structures, an α-helix in 16 structures and turns in four structures in the NMR ensemble) and Val 120-Trp 123 (α-helix in 33 structures and turns in 17 structures); no hydrogen bond restraints were included for these residues in the structure calculations, but both these regions form short 310-helices in the X-ray structure. Interestingly, recent MD simulations of native hen lysozyme in solution and crystal environments show in both of these regions higher populations of CO(i)-NH(i+4) α-helical hydrogen bonds than the CO(i)NH(i+3) hydrogen bonds expected for a 310-helix (Stocker et al. 2000). This behavior, and the results from the NMR structure calculations, may reflect the similarity in the energy of α- and 310-helices for these residues in hen lysozyme.
Figure 1a ▶ shows the backbone RMSD relative to the mean structure for each residue in the protein. Some disorder in the family of structures remains in four regions of the sequence, involving particularly Gly 22, Thr 47 and Asp 48, Arg 68 to Gly 71, and Gly 102 (backbone RMSD to the mean structure greater than 0.9Å for these residues). All these regions include residues that have a significant solvent accessibility for the polypeptide backbone (main-chain accessibility >70% for Gly 22, Thr 47, Gly 71, Gly 102). Although experimental 15N relaxation measurements for the main-chain amide groups of hen lysozyme (Buck et al. 1995) show that most residues have order parameter values greater than 0.8, order parameters in the range 0.7 to 0.8 are seen for Arg 45, Thr 47, Asp 48, Arg 68, Thr 69, Gly 71, Ser 72, Cys 115, Thr 118, Cys 127, Arg 128, and Leu 129. In addition, Ser 85, Gly 102, Asn 103, and Gly 104 have order parameters less than 0.7. Thus six of the eight residues with significant disorder in the NMR ensemble have backbone order parameters less than 0.8. Furthermore, an elevated T1/T2 ratio of 4.18 for Gly 22 (the mean ratio for hen lysozyme is 3.32±0.13) indicates that there are motions on the microsecond to millisecond timescale for this residue (Buck et al. 1995). Therefore, these data suggest that at least some of the disorder in the NMR ensemble reflects the presence of internal motions in the protein in solution rather than merely a lack of NMR restraints.
A comparison of the backbone RMSD for the average coordinates of the set of NMR structures from the X-ray structure of lysozyme is shown in Figure 1b ▶. Deviations are seen for the four regions that have some disorder in the family of NMR structures. Interestingly, residues in three of these regions also have the highest main-chain crystallographic temperature factors (excluding the C terminus) in the X-ray structure (Fig. 1c ▶). Temperature factors greater than 20 Å2 are seen for the amide N atoms of Thr 47, Asp 48, Pro 70-Arg 73, and Asp 101-Asn 103 (also Arg 125-Leu 129). Furthermore, comparisons of 33 crystal structures of hen lysozyme show variations between the structures in the main-chain conformation for Pro 70-Ser 72 and reveal that the orientation of the turn between the first two strands of the β-sheet (particularly Thr 47-Gly 49) may be affected by crystal contacts (C. Redfield, unpubl.). The fact that there are some differences between the solution and crystal structures of the protein in these regions is consistent with this observation. Gly 22 is incorporated in a β-bridge motif in the X-ray structure, involving Tyr 20 and Tyr 23, and does not have elevated main-chain B factors. This β-bridge is also present in 13 of the 50 NMR structures; these structures show much lower backbone RMSD values to the X-ray structure for Gly 22 (∼1.2Å) than does the mean NMR structure (2.7Å). The ensemble of NMR conformers therefore includes the turn orientation found in the X-ray structure, along with a variety of other orientations. This is consistent with the evidence from 15N relaxation studies for motions involving Gly 22 on a microsecond to millisecond timescale in solution.
A significant deviation between the average NMR structure and the X-ray structure is also seen for Lys 116-Thr 118, a region that is not found to be disordered in the ensemble of NMR structures. Gly 117 has, however, the highest main-chain solvent accessibility (97%) of any residue in the protein, and there are no long range NOEs (i,i +5 or greater) identified for these residues. Consequently, the NMR data set may not be sufficient to define the correct conformation of the protein in this region; despite this lack of NOEs, the absence of significant disorder within the NMR ensemble probably reflects the close proximity of these residues to the restraints provided by the Cys 30-Cys115 disulphide bridge. It is interesting in this respect, however, that MD simulations of native hen lysozyme showed a significant rearrangement of residues Cys 115-Asp 119 during 2ns simulations in solution and crystal environments (Stocker et al. 2000). In addition, analysis of 15N T1/T2 ratios using an anisotropic rotational diffusion model shows poor agreement with T1/T2 values predicted from the X-ray structure for Thr 118 (C. Redfield, unpubl.).
Assessing the importance of the dipolar coupling restraints in the NMR structure determination
As described in the previous section, the quality of the NMR structure of hen lysozyme reported here is significantly improved compared with the structure reported in 1993. This difference reflects the enlarged data set of NOE and dihedral angle restraints, improvements to the force field used in the structure calculations, and the inclusion of the dipolar coupling data. To identify separately the effects that result from adding the dipolar couplings, the final ensemble of structures of hen lysozyme (structure set 1) has been compared with an ensemble of 50 low-energy structures calculated using the same refinement protocol but excluding the residual dipolar coupling data (structure set 2).
The inclusion of the dipolar coupling data gives an overall increased precision in the definition of the structure (average backbone RMSD to mean 0.50±0.13 Å for set 1 compared with 0.60±0.14 Å for set 2) and a closer similarity to the X-ray structure (average backbone RMSD to X-ray structure 1.49±0.10 Å for set 1 compared with 1.69±0.12 Å for set 2). The addition of the dipolar coupling data also results in structures with improved stereochemical quality (for structures in set 1, 74.2±2.0% of the φ,ψ; torsion angles lie in the most favored region of the Ramachandran plot compared with 65.9±2.6% for structures in set 2).
The definition of the relative orientations of the two structural domains in the protein is very substantially improved for the structures in sets 1 and 2 compared with the definition in the structures reported previously. To characterize this, we have determined the angle between helix C in the α-domain and the first strand of the β-sheet in the β-domain. This helix-strand angle has a value of 48.7° in the X-ray structure. In the structures reported previously, this angle is 34.3±13.4°, whereas it has values of 52.6±4.0° and 53.6±5.6° in structure sets 1 and 2, respectively. The better definition of the helix-strand angle and the closer agreement with the value observed in the X-ray structure for the structures in set 1 reflects both the additional NOEs from the heteronuclear spectra (82 inter-domain NOEs compared with 45 in the 1993 data set) and the dipolar coupling data. The improved definition of the domain orientations is further shown by the relative values of the backbone RMSD from the mean structure for residues in the β-domain of the protein when the structures are superimposed using the full sequence (0.58±0.19Å for set 1, 0.74±0.25Å for set 2, and 2.23±0.40Å for the 1993 structures) or only the α-domain residues (0.73±0.2Å for set 1, 0.91±0.30Å for set 2, and 2.63±0.49Å for the 1993 structures).
Cornilescu et al. (1998) have shown that the quality of NMR structures can be assessed by comparison of predicted NMR parameters with experimental NMR data that were not used in the refinement process. They have introduced a quality, Q, factor defined as
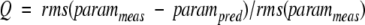 |
in which param is a measurable NMR parameter such as a residual dipolar coupling or orientation-induced chemical shift change. Here we have used this approach to compare the lysozyme solution structures calculated with and without the two sets of residual dipolar couplings. The additional data used to determine the Q factor are, firstly, orientation-induced 15N chemical shift changes measured for the 5% and 7.5% bicelle solutions (Boyd and Redfield, 1999); secondly, a set of residual dipolar couplings measured for lysozyme in a bicelle mixture composed of 3.8% DTDPC: DHPC:CTAB:DMPE-DTPA:La+3 (3.0: 1.0: 0.4: 0.07: 0.06; J.Boyd and C. Redfield, unpubl.); and thirdly, T1/T2 values obtained from 15N relaxation measurements (Buck et al. 1995). The Q factors obtained with the 15N chemical shift changes are 0.36 +/−0.01 and 0.75 +/−0.03 for the structures in sets 1 and 2 obtained with and without the residual dipolar couplings, respectively. The Q factors obtained with the additional set of dipolar couplings are 0.30 +/−0.01 and 0.65 +/−0.03 for the structures in sets 1 and 2, respectively. The Q factors obtained with the T1/T2 ratios are 0.030 +/−0.001 and 0.0455 +/−0.001 for the structures in sets 1 and 2, respectively. Thus in all three cases the Q values are substantially lower for the structures refined using the dipolar couplings than for those refined without these data. The agreement between the experimental T1/T2 values and those calculated using the structures refined with dipolar couplings is comparable to that obtained with the X-ray structure. The average anisotropy (D∥/D⊥) of 1.26±0.01 obtained from the family of 50 structures in set 1 is the same as that found for the X-ray structure. The structures refined without the dipolar couplings give significantly worse agreement with the T1/T2 data with an average anisotropy of only 1.18±0.02.
Figure 6 ▶ compares the variation along the protein sequence in backbone RMSD values with the mean structure, and between the mean and X-ray structures for the two sets of structures. The improvements for set 1 on the inclusion of the dipolar couplings are concentrated around residues Asn 46-Gly 49, Arg 68-Pro 70, and Ala 82-Leu 84. These are the three areas in which particularly large deviations are seen in the ensemble of NMR structures reported previously (Smith et al. 1993). The changes for residues Ala 82-Leu 84 on the addition of the dipolar coupling data are particularly interesting. Residues Cys 80-Leu 84 form a 310-helix in the X-ray structure, but the conformation of these residues is disordered in the ensemble of structures reported previously (Smith et al. 1993). Despite the addition of NOEs from the heteronuclear spectra, such as Ser 81 Hα-Leu 84 Hβ1, which help in the definition of this region, significant deviations from the X-ray structure are still seen for the NMR structures in set 2 (Fig. 6b ▶). The dipolar coupling data set includes two restraints for the NH bond vectors of each of the residues in the range Cys 80-Leu 84. The inclusion of these data leads to the substantial increase in the similarity of the NMR and X-ray structures in this region (e.g., RMSD between the mean and X-ray structure for Leu 83 is reduced from 4.24 Å for set 2 to 2.03 Å for set 1).
Fig. 6.

Comparison of (a) the average backbone RMSD values (in Å) from the mean structure and (b) the backbone RMSD values (in Å) between the mean structure and the X-ray structure for the ensembles of structures in sets 1 and 2. In each case the RMSD values are shown as a function of residue number, (filled circles) the 50 structures in set 1, calculated including the dipolar coupling data, (open circles) the 50 structures in set 2, calculated without the dipolar coupling data.
There is also a significant difference between the two sets of NMR structures for the region surrounding Gly 22, a larger RMSD to the mean structure being seen for set 1 (0.95 Å for Gly 22) than set 2 (0.23 Å for Gly 22). As discussed above, a β-bridge involving Tyr 20 and Tyr 23, which is also defined in the X-ray structure, is present in 13 of the 50 structures of set 1. This β-bridge is not present in any of the structures in set 2; in this case, therefore, the addition of the dipolar coupling data enables the β-bridge to be defined. However, there is an incompatibility between the dipolar coupling of Gly 22 measured in the 5% bicelle solution and an NOE observed between Tyr 23 HN and Trp 28 HZ2. In the structures in which the β-bridge is defined, there is close agreement between the calculated and experimental dipolar couplings of Gly 22 (difference <0.5 Hz), but the NOE is violated by 0.3–0.4 Å. In contrast, in the structures in which there is no β-bridge, there is a violation of 2.3–4.1 Hz for the experimental dipolar coupling of Gly 22, but there are no violations of the Tyr 23 HN-Trp 28 HZ2 NOE greater than 0.3Å. This incompatibility between the data presumably reflects the mobility of this region in solution and means that the β-bridge is not the only conformation that is observed in the ensemble of set 1 structures. This observation suggests that a simple comparison of RMSD values may not be a definitive measure of the quality of the structure in solution where motional effects are significant, and the average structure does not represent the ensemble of contributing conformers in a meaningful way.
Interestingly, the β-strand secondary structure in the triple stranded antiparallel β-sheet is better defined in the structures in set 2 than those in set 1. The first two strands are defined in all 50 of the structures in set 2, whereas in set 1 these full strands are only defined in 32 structures and there is a β-bridge in 14 structures. In addition, a β-bridge involving Asn 59 in the third strand is defined in 25 of the structures of set 2 but this is missing in all the structures in set 1. A closer analysis of the structures shows that this difference does not reflect large structural changes in this region but, instead, slight alterations in the relative orientations of the amide and carbonyl pairs for 54NH-42CO and 58NH-53CO because of the dipolar coupling restraints. These alterations result in the hydrogen bonds no longer being identified by the Kabsch and Sander method, but do not result in significant violations of the hydrogen bond restraints as only weak restraints of this type (NH(i) to O(i-4) 1.3–2.3Å and N(i) to O(i-4) 2.3–3.3Å) are included in the structure calculations.
Conclusions
In most cases in which NMR techniques are used to determine the structure of a globular protein, the close packed interior core of the protein is defined with a high precision in the ensemble of calculated structures (Doreleijers et al. 1998). The good definition reflects in general the considerable number of NOE distance restraints that have been identified for the core hydrophobic residues. This is the case for the ensemble of lysozyme structures determined here in which there is a heavy atom RMSD of 0.57±0.17Å from the mean structure for the 39 residues which have a solvent accessibility less that 10%. For understanding the biological role of a protein, the areas of most interest are often, however, exposed surface regions such as the active sites of enzymes or the binding sites of proteins involved in intermolecular association. Here, the NMR ensemble will often be less well defined. In some cases this reflects the high mobility of surface residues resulting in averaging of the NMR parameters so that they do not correspond to a single conformer. However, even when there is a single preferred conformation, there may be difficulties in collecting sufficient NOE data to define the structure adequately in exposed regions as there may not be many atoms in close proximity of a given proton. In such a situation, other types of NMR data, particularly those that give restraints of a longer range or different nature, can play a very important role in increasing the accuracy of an NMR structure determination. In this work we have investigated this for hen lysozyme by examining the effects of including in the structure calculations a relatively small number of dipolar coupling restraints (209 out of a total of 2011 experimental restraints). These additional restraints have improved the quality of the structure (improved stereochemical quality, reduced Q factors, and reduced RMSD to the X-ray structure). The refinement has resulted in an ensemble of structure in which the backbone RMSD values are 0.43±0.10Å from the mean NMR structure and 1.32±0.08Å from the X-ray structure when 12 residues in the mobile regions of the protein are excluded from the RMSD calculations (Table 2).
Interestingly, however, there is still conformational disorder within the NMR ensemble in some regions of the structure, most notably in the long loop and involving residues in the turns between helices A and B and between the first two strands in the β-sheet. Some significant differences also remain between the NMR and X-ray structures of hen lysozyme (Fig. 1, 6 ▶ ▶). By comparison with results from experimental 15N relaxation studies (Buck et al. 1995) and theoretical MD simulations (Stocker et al. 2000), we have been able to establish that for all these regions of difference or disorder there is evidence for motions, either on a fast picosecond or on a slower microsecond to millisecond timescale, within the protein in solution. For lysozyme, in most regions of the sequence the X-ray structure provides a reasonably good representation of the average structure in solution. However, for Pro 70-Ser 72 significant differences are observed between different crystal structures of the protein, and for Thr 47-Gly 49 the conformation in X-ray structures is affected by crystal contacts (C. Redfield, unpubl.). For these regions in which differences are observed between the X-ray and solution structures, the NMR ensemble reported here may therefore provide a more realistic structural representation. More importantly, however, the ensemble of NMR structures may give information about the range of conformations that are present for lysozyme in solution. For the highly mobile regions of the protein a single structure defined either by NMR or by X-ray diffraction methods cannot provide a full description of the protein conformation. Moreover, in some cases conformers present only in low populations may play an important role in the dynamics, folding, or function of the protein. The availability of a variety of conformations for lysozyme consistent with the NMR data provides insight into the conformations likely to be accessible in solution.
Materials and methods
Protein samples
Hen egg white lysozyme was expressed in Aspergillus niger and purified from filtered culture medium as described previously (MacKenzie et al. 1996). For the 15N-labeled sample, 15NH4Cl was used as the sole nitrogen source. For the double-labeled 15N,13C sample, 15NH4Cl was used as the sole nitrogen source and 13C-labeled glucose as the sole carbon source. The 15N NMR sample was prepared to contain ca. 4mM protein in 95%H2O/5%D2O. The 15N,13C NMR samples contained protein concentrations of ca. 1.5mM, the protein being dissolved in either 90%H2O/10%D2O or 100%D2O. All samples were at pH 3.8 unless otherwise stated, and the NMR experiments were performed at 35°C.
NMR experiments and data analysis
NMR spectra were recorded on home-built spectrometers at the Oxford Centre for Molecular Sciences with 1H operating frequencies of 500 MHz and 600 and 750 MHz and on a Bruker DMX spectrometer at the Institute of Organic Chemistry, University of Frankfurt, with a 1H operating frequency of 600 MHz. The spectrometers at the Oxford Centre for Molecular Sciences are equipped with Oxford Instruments Company magnets (Oxford, UK), OMEGA software, and digital control equipment (Bruker Instruments), home-built triple resonance pulsed field gradient probe heads and home-built linear amplifiers. Data processing was performed using Felix 2.3 from MSI (San Diego, CA).
The HMQCJ spectrum (Kay and Bax 1990) was used to determine 3J(HN,Hα) coupling constant values. The coupling constants were fitted to doublets in ω1 by optimization of the coupling constant and line widths for each of the doublet components using home-written software. For the soft HCCH-COSY and HCCH-E.COSY spectra (Eggenberger et al. 1992; Karimi-Nejad et al. 1994) mirror-image linear prediction as implemented in Felix97 was performed to enhance resolution in ω2 (Zhu and Bax 1990). Quantitative 3J(Hβ,C′), and 3J(Hα,Hβ) coupling constants were extracted from the spectra using published methods (Schwalbe et al. 1994).
NOE intensities in the heteronuclear 3D NOESY-HMQC spectra were estimated from peak heights, the cross peaks being divided into four categories (strong, medium, weak or very weak) equivalent to those used previously (Smith et al. 1993). The corresponding distance restraints used for the four categories were 1.8–2.5Å (strong), 1.8–3.0Å (medium), 1.8–4.5Å (weak), and 1.8–5.5Å (very weak). The NOE data set also contained some restraints from cross peaks that are only observed in 1H NOESY spectrum recorded with a long 500 ms mixing time. For these cross peaks, the corresponding distance restraint was 1.8–7.5Å (Smith et al. 1993). Pseudoatoms were used in the structure calculations where no stereospecific assignments had been achieved, the necessary corrections being made to the distance range (Wüthrich et al 1983). In addition, 0.5Å was added to the upper limit of distance restraints when the NOE involved protons of a methyl group (Wüthrich, 1986).
3J(HN,Hα) coupling constants were used to give φ torsion angle restraints. A restraint of −60±30° was used for residues in helices with a 3J(HN,Hα) value less than 5.5 Hz, whereas a restraint of −120±40° was used for residues with a 3J(HN,Hα) value greater than 8.5 Hz. For residues in which the χ1 rotamer had been identified on the basis of 3J(Hα,Hβ) and 3J(Hβ,C′), coupling constant values, χ1 restraints of -60±30°, 60±30°, or 180±30° were used. Hydrogen bond restraints between NH(i) and O(i-4) (1.3–2.3Å) and between N(i) and O(i-4) (2.3–3.3Å) were included for residues in the helical regions Cys 6-His 15, Leu 25-Asn 37, and Thr 89-Asp 101 in which there are slow amide proton exchange rates. For the β-sheet, hydrogen bond restraints were included between the pairs Ala 42-Gly 54, Arg 44-Asp 52, Asn 46-Ser 50, and Tyr 53-Ile 58 in which slow amide proton exchange rates are observed experimentally.
Measurement of residual dipolar couplings
Residual dipolar couplings were measured for 0.5mM 15N-labeled hen lysozyme in two different bicelle solutions. The first contained 5% w/v DMPC and DHPC, q = 2.9, in 10mM phosphate buffer at pH 6.5 (93%/7% H2O/D2O) with a small amount of dioxan as a 1H chemical shift reference. The second solution contained 7.5% w/v DMPC and DHPC but with CTAB added to give the bicelles an overall positive charge (DMPC:DHPC:CTAB = 2.9: 1.0: 0.1). The sample preparation protocol followed that described by Ottiger and Bax (1998).
The 1H-15N residual dipolar couplings were measured from the 1JNH splitting appearing in the 15N dimension of an HSQC experiment that incorporated an S3E pulse sequence element and could be used to select for either multiplet component in separate experiments (Meissner et al. 1997). The experiments were recorded at 750 MHz using 128 (t1–15N) and 2048 (t2–1H) complex points with acquisition times of 28.5 and 97.3 ms, respectively. The data were processed to give a final digital resolution of 1 Hz/pt (F1) and 3 Hz/pt (F2). Pairs of spectra were collected at 35°C for the 5% DMPC:DHPC and 7.5% DMPC:DHPC:CTAB bicelle solutions. A pair of spectra was also collected at 35°C for an isotropic solution of lysozyme dissolved in 10mM phosphate buffer at pH 6.5. The residual dipolar coupling was taken as the difference between the splitting observed in the oriented bicelle and isotropic bicelle solutions (1DNH = 1JNH(bic) − 1JNH(iso)); 1JHN is assumed to be negative.
Structure calculations and analysis
Structures were calculated using XPLOR version 3.8 (Brünger 1992) and the PARALLDG5.1 force field of Linge and Nilges (1999). A simulated annealing protocol starting from randomized coordinate positions was used (Nilges et al. 1988; Wiles et al. 1997) for the first stages of the structure calculations in which the NOE, dihedral angle, and hydrogen bond restraints were included but not the dipolar coupling restraints. This initial simulated annealing consisted of 30000 steps at 1000K followed by cooling to 100K over 15000 steps. This was followed by a refinement stage consisting of 2000 steps at 2000K and 2000 steps during cooling to 100K. The 15 lowest energy structures from this protocol were selected for a further refinement in which the dipolar coupling data (along with the NOE, dihedral angle, and hydrogen bond restraints) were included.
The dipolar coupling refinement used a version of XPLOR modified to include dipolar coupling restraints (Clore et al. 1998b). A simulated annealing protocol was used that had an initial temperature of 1500K with 20000 cooling steps down to a temperature of 100K. A harmonic potential was used for the dipolar coupling restraints with final force constants in the protocol of 0.3 kcal mol−1 Hz−2 and 0.5 kcal mol−1 Hz−2 for the 5% DMPC:DHPC (5% bicelles) and 7.5% DMPC:DHPC:CTAB (7.5% bicelles) data, respectively. The inclusion of the dipolar restraints in the structure calculations requires values of the parameters Da, the axial component of the alignment tensor, and R, the rhombicity, to be defined. Estimates of these were obtained for the two sets of dipolar couplings using the approach of Clore et al. (1998a) (Da = 15.1, R = 0.34 for the 5% bicelle data; Da = 12.2, R = 0.16 for the 7.5% bicelle data). Preliminary calculations (25 sets of 15 structures) were then run to optimize the values of these parameters. Including only the dipolar restraints from the 5% bicelle data, R values in the range 0.28–0.38 in 0.02 steps and Da values varying from 14.5–16.0 in 0.25 steps were used. Similarly, calculations with only the 7.5% bicelle data being included were run with R in the range 0.10–0.20 and Da in the range 11.5–14.0. In each case, the values of these parameters that gave the lowest energy structures were selected (for 5% bicelle data Da = 15.5, R = 0.32; for 7.5% bicelle data Da = 13.5, R = 0.17; Clore et al. 1998b). These parameters were used in calculations in which 20 structures were calculated from each of the 15 structures from the initial protocol. The 50 lowest energy structures were selected for analysis (structure set 1). For comparison another set of 50 structures was calculated using an identical protocol but excluding the dipolar coupling data (structure set 2).
The stereochemical quality of the structures was analysed using the program PROCHECK (Laskowski et al. 1993), and regions of secondary structure were identified using the program DSSP (Kabsch and Sander 1983). Solvent accessible surface areas were calculated for the structures using the program NACCESS (Hubbard and Thornton 1993).
Acknowledgments
This is a contribution from the Oxford Centre for Molecular Sciences, which is supported by the U.K. Biotechnology and Biological Sciences Research Council (BBSRC), the Engineering and Physical Sciences Research Council, and the Medical Research Council. The research of C.M.D. is supported in part by a program grant from the Wellcome Trust and by an International Research Scholars award from the Howard Hughes Medical Research Institute. L.J.S. is a Royal Society Research Fellow. C.R. is a BBSRC Advanced Research Fellow. M.B. has a NRSA postdoctoral fellowship from NIH. H.S. is supported by the Massachusetts Institute of Technology and the Karl-Winnacker Foundation. Some of the NMR experiments were performed at the Large Scale Facility at the University of Frankfurt/M. We thank Marius Clore for providing the version of XPLOR modified to include dipolar coupling restraints.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CTAB, cetyltrimethylammonium bromide
DHPC, dihexanoyl-phosphatidylcholine
DMPC, dimyristoyl-phosphatidlycholine
E.COSY, exclusion correlation spectroscopy
HMQC, heteronuclear multiple-quantum correlation
HSQC, heteronuclear single-quantum correlation
MD, molecular dynamics
NMR, nuclear magnetic resonance
NOE, nuclear Overhauser enhancement
NOESY, nuclear Overhauser enhancement and exchange spectroscopy
RMSD, root-mean-square difference
TOCSY, total correlation spectroscopy
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.43301.
References
- Bartik, K. and Redfield, C. 1993. A method for the estimation of chi-1 torsion angles in proteins. J. Biomol. NMR 3 415–428. [Google Scholar]
- Bax, A. and Tjandra, N. 1997. High resolution heteronuclear NMR of human ubiquitin in an aqueous liquid crystalline medium. J. Biomol. NMR 10 289–292. [DOI] [PubMed] [Google Scholar]
- Boyd, J. and Redfield, C. 1999. Characterization of 15N chemical shift anisotropy from orientation-dependent changes to 15N chemical shifts in dilute bicelle solutions. J. Amer. Chem. Soc. 121 7441–7442. [Google Scholar]
- Brünger, A.T. 1992. XPLOR Version 3.1: A system for X-ray crystallography and NMR. Yale University Press, New Haven CT.
- Buck, M., Boyd, J., Redfield, C., MacKenzie, D.A., Jeenes, D.J., Archer, D.B., and Dobson, C.M. 1995. Structural determinants of protein dynamics: Analysis of 15N NMR relaxation measurements for main chain and side chain nuclei of hen egg white lysozyme. Biochemistry 34 4041–4055. [DOI] [PubMed] [Google Scholar]
- Case, D.A. 1999. Calculations of NMR dipolar coupling strengths in model peptides. J. Biomol. NMR 15 95–102. [DOI] [PubMed] [Google Scholar]
- Clore, G.M. and Gronenborn, A.M. 1998. New methods of structure refinement for macromolecular structure determination by NMR. Proc. Natl. Acad. Sci. 95 5891–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., and Bax, A. 1998a. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J. Magn. Reson. 133 216–221. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., and Tjandra, N. 1998b. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson. 131 159–162. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., Marquardt, J.L., Ottiger, M., and Bax, A. 1998. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Amer. Chem. Soc. 120 6836–6837. [Google Scholar]
- de Alba, E., De Vries L., Farquhar, M.G., and Tjandra, N. 1999. Solution structure of human GAIP (Gα interacting protein): A regulator of G protein signaling. J. Mol. Biol. 29 927–939. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M., Evans, P.A., and Radford, S.E. 1994. Understanding how proteins fold—the lysozyme story so far. Trends Biochem. Sci. 19 31–37. [DOI] [PubMed] [Google Scholar]
- Doreleijers, J.F., Rullmann, J.A.C., and Kaptein, R. 1998. Quality assessment of NMR structures: A statistical survey. J. Mol. Biol. 281 149–164. [DOI] [PubMed] [Google Scholar]
- Driscoll, P.C., Clore, G.M., Marion, D., Wingfield, P.T., and Gronenborn, A.M. 1990. Complete resonance assignment for the polypeptide backbone of interleukin 1β using three-dimensional heteronuclear NMR spectroscopy. Biochemistry 29 3542–3556. [DOI] [PubMed] [Google Scholar]
- Eggenberger, U., Kariminejad, Y., Thuring, H., Ruterjans, H., and Griesinger, C. 1992. Determination of H-alpha,H-beta and H-beta,C′ coupling-constants in C-13 labeled proteins. J. Biomol. NMR 2 583–590. [Google Scholar]
- Fesik, S.W., Eaton, H.L., Olejniczak, E.T., Zuiderweg, E.R.P., Mcintosh, L.P., and Dahlquist, F.W. 1990. 2D and 3D NMR spectroscopy employing C-13-C-13 magnetization transfer by isotropic mixing—spin system identification in large proteins. J. Amer. Chem. Soc. 112 886–888. [Google Scholar]
- Hubbard, S.J. and Thornton, J.M. 1993. NACCESS computer program, Departments of Biochemistry and Molecular Biology, University College, London.
- Ikura, M., Kay, L., Tschudin, R., and Bax, A. 1990. 3-dimensional NOESY-HMQC spectroscopy of a C-13-labeled protein. J. Magn. Reson. 86 204–209. [Google Scholar]
- Johnson, L.N., Cheetham, J., McLaughlin, P.J., Acharya, K., Barford, D., and Phillips, D.C. 1988. Protein oligosaccharide interactions—lysozyme, phosphorylase, amylases. Curr. Top. Microbiol. 139 81–134. [DOI] [PubMed] [Google Scholar]
- Jollès, P. and Jollès, J. 1984. What's new in lysozyme research—always a model system, today as yesterday? Mol. Cell Biochem. 63 165–189. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure—pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Karimi-Nejad, Y., Schmidt, J.M., Rüterjans, H., Schwalbe, H., and Griesinger, C. 1994. Conformation of valine side chains in Ribonuclease T1 determined by NMR studies of homonuclear and heteronuclear 3J coupling constants. Biochemistry 33 5481–5492. [DOI] [PubMed] [Google Scholar]
- Kay, L.E. and Bax, A. 1990. New methods for the measurement of NH-CαH coupling constants in N-15-labeled proteins. J. Magn. Reson. 86 110–126. [Google Scholar]
- Kay, L.E., Xu, G.Y., Singer, A.U., Muhandiram, D.R., and Forman-Kay, J.D. 1993. A gradient-enhanced HCCH TOCSY experiment for recording side-chain H-1 and C-13 correlations in H2O samples of proteins. J. Magn. Reson. Ser. B 101 333–337. [Google Scholar]
- Kraulis, P.J. 1991. Molscript: A program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24 946–950. [Google Scholar]
- Krebs, M.R.H., Wilkins, D.K., Chung, E.W., Pitkeathly, M.C., Chamberlain, A.K., Zurdo, J., Robinson, C.V., and Dobson, C.M. 2000. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the β-domain. J. Mol. Biol. 300 541–549. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Linge, J.P. and Nilges, M. 1999. Influence of non-bonded parameters on the quality of NMR structures: A new force field for NMR structure calculation. J. Biomol. NMR 13 51–59. [DOI] [PubMed] [Google Scholar]
- Losonczi, J.A. and Prestegard, J.H. 1998. Improved dilute bicelle solutions for high resolution NMR of biological macromolecules. J. Biomol. NMR 12 447–451. [DOI] [PubMed] [Google Scholar]
- MacKenzie, D.A., Spencer, J.A., Le Gal-Coeffet, M.F., and Archer, D.B. 1996. Efficient production from Aspergillus niger of a heterologous protein and an individual protein domain, heavy isotope labelled, for structure, function analysis. J. Biotechnol. 46 85–93. [DOI] [PubMed] [Google Scholar]
- Markus, M.A., Gerstner, R.B., Draper, D.E., and Torchia, D.A. 1999. Refining the overall structure and subdomain orientation of ribosomal protein S4 Δ41 with dipolar couplings measured by NMR in uniaxial liquid crystalline phases. J. Mol. Biol. 292 375–387. [DOI] [PubMed] [Google Scholar]
- Meissner, A., Duus, J.O., and Sorensen, O.W. 1997. Integration of spin-state-selective excitation into 2D NMR correlation experiments with heteronuclear ZQ/2Q pi rotations for 1J(XH)-resolved E.COSY-type measurement of heteronuclear coupling constants in proteins. J. Biomol. NMR 10 89–94. [DOI] [PubMed] [Google Scholar]
- Nilges, M., Gronenborn, A.M., Brunger, A.T., and Clore, G.M. 1988. Determination of 3-dimensional structures of proteins by simulated annealing with interproton distance restraints—application to crambin, potato carboxypeptidase inhibitor and barley serine proteinase inhibitor-2. Protein Eng. 2 27–38. [DOI] [PubMed] [Google Scholar]
- Ottiger, M and Bax, A. 1998. Characterization of magnetically oriented phospholipid micelles for measurement of dipolar couplings in macromolecules. J. Biomol. NMR 12 361–372. [DOI] [PubMed] [Google Scholar]
- Prestegard, J.H. 1998. New techniques in structural NMR—anisotropic interactions. Nat. Struct. Biol. 5 Suppl: 517–522. [DOI] [PubMed] [Google Scholar]
- Ramirez, B.E. and Bax, A. 1998. Modulation of the alignment tensor of macromolecules dissolved in a dilute liquid crystalline medium. J. Amer. Chem. Soc. 120 9106–9107. [Google Scholar]
- Redfield, C. and Dobson, C.M. 1988. Sequential 1H NMR assignments and secondary structure of hen egg white lysozyme in solution. Biochemistry 27 122–136. [DOI] [PubMed] [Google Scholar]
- Sass, J., Cordier, F., Hoffmann, A., Cousin, A., Omichinski, J.G., Lowen, H., and Grzesiek, S. 1999. Purple membrane induced alignment of biological macromolecules in the magnetic field. J. Amer. Chem. Soc. 121 2047–2055. [Google Scholar]
- Sattler, M., Schwendinger, M.G., Schleucher, J., and Griesinger, C. 1995. Novel strategies for sensitivity enhancement in heteronuclear multidimensional NMR experiments employing pulsed-field gradients. J. Biomol. NMR 6 11–22. [DOI] [PubMed] [Google Scholar]
- Schwalbe, H., Marino, J.P., King, G.C., Wechselberger, R., Bermel, W., and Griesinger C. 1994. Determination of a complete set of coupling constants in C-13-labeled oligonucleotides. J. Biomol. NMR 4 631–644. [DOI] [PubMed] [Google Scholar]
- Smith, L.J., Sutcliffe, M.J., Redfield, C., and Dobson, C.M. 1991. Analysis of φ and χ 1 torsion angles for hen lysozyme in solution from 1H NMR spin, spin coupling constants. Biochemistry 30 986–996. [DOI] [PubMed] [Google Scholar]
- Smith, L.J., Sutcliffe, M.J., Redfield, C., and Dobson, C.M. 1993. Structure of hen lysozyme in solution. J. Mol. Biol. 229 930–944. [DOI] [PubMed] [Google Scholar]
- Smith, L.J., Mark, A.E., Dobson, C.M., and van Gunsteren, W.F. 1995. Comparison of MD simulations and NMR experiments for hen lysozyme—analysis of local fluctuations, cooperative motions, and global changes. Biochemistry 34 10918–10931. [DOI] [PubMed] [Google Scholar]
- Stocker, U., Spiegel, K., and van Gunsteren, W.F. 2000. On the similarity of properties in solution or crystalline state: a molecular dynamics study of hen lysozyme. J. Biomol. NMR 18 1–12. [DOI] [PubMed] [Google Scholar]
- Tjandra, N. and Bax, A. 1997. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278 1111–1114. [DOI] [PubMed] [Google Scholar]
- Tjandra, N., Omichinski, J.G., Gronenborn, A.M., Clore, G.M., and Bax, A. 1997. Use of dipolar 1H–15N and 1H–13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Biol. 4 732–738. [DOI] [PubMed] [Google Scholar]
- Tsui, V., Zhu, L., Huang, T.H., Wright, P.E., and Case, D.A. 2000. Assessment of zinc finger orientations by residual dipolar coupling constants. J. Biomol. NMR 16 9–21. [DOI] [PubMed] [Google Scholar]
- Vaney, M.C., Maignan, S., RiesKautt, M., and Ducruix, A. 1996. High-resolution structure (1.33 angstrom) of a HEW lysozyme tetragonal crystal grown in the APCF apparatus. Data and structural comparison with a crystal grown under microgravity from SpaceHab-01 mission. Acta. Crystallogr. D 52 505–517. [DOI] [PubMed] [Google Scholar]
- Wiles, A.P., Shaw, G., Bright, J., Perczel, A., Campbell, I.D., and Barlow, P.N. 1997. NMR studies of a viral protein that mimics the regulators of complement activation. J. Mol. Biol. 272 253–265. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of Protein and Nucleic Acids, John Wiley, New York.
- Wüthrich, K., Billeter, M., and Braun, W. 1983. Pseudo-structures for the 20 common amino-acids for use in studies of protein conformations by measurements of intramolecular proton proton distance constraints with nuclear magnetic-resonance. J. Mol. Biol. 169 949–961. [DOI] [PubMed] [Google Scholar]
- Zhu, G. and Bax, A. 1990. Improved linear prediction for truncated signals of known phase. J. Magn. Reson. 90 405–410. [Google Scholar]
- Zweckstetter, M. and Bax, A. 2000. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J. Amer. Chem. Soc. 122 3791–3792. [Google Scholar]