

The structure of HsaD, a carbon–carbon bond serine hydrolase involved in steroid catabolism that is critical for the survival of M. tuberculosis inside human macrophages, has been solved by X-ray crystallography. Data were collected at the Diamond Light Source in Oxfordshire, England: this paper describes one of the first structures determined at the new synchrotron.
Keywords: HsaD, MCP hydrolases, C–C bond hydrolases, cholesterol, tuberculosis, Diamond
Abstract
Tuberculosis is a major cause of death worldwide. Understanding of the pathogenicity of Mycobacterium tuberculosis has been advanced by gene analysis and has led to the identification of genes that are important for intracellular survival in macrophages. One of these genes encodes HsaD, a meta-cleavage product (MCP) hydrolase that catalyzes the hydrolytic cleavage of a carbon–carbon bond in cholesterol metabolism. This paper describes the production of HsaD as a recombinant protein and, following crystallization, the determination of its three-dimensional structure to 2.35 Å resolution by X-ray crystallography at the Diamond Light Source in Oxfordshire, England. To the authors’ knowledge, this study constitutes the first report of a structure determined at the new synchrotron facility. The volume of the active-site cleft of the HsaD enzyme is more than double the corresponding active-site volumes of related MCP hydrolases involved in the catabolism of aromatic compounds, consistent with the specificity of HsaD for steroids such as cholesterol. Knowledge of the structure of the enzyme facilitates the design of inhibitors.
1. Introduction
HsaD is a member of the αβ-hydrolase superfamily, which includes the meta-cleavage product (MCP) hydrolases (Seah et al., 2007 ▶). MCP hydrolases occur in the microbial pathways responsible for the aerobic catabolism of aromatic compounds, catalysing the hydrolytic cleavage of a carbon–carbon bond in the 2-hydroxy-6-oxo-dienoates that result from the meta cleavage of catechols by extradiol dioxygenases. HsaD from Mycobacterium tuberculosis H37Rv is a class I MCP hydrolase; this class includes enzymes involved in steroid and biphenyl catabolism (Seah et al., 2007 ▶). Recent work has demonstrated that HsaD has high specificity for the steroid MCP 4,5-9,10-diseco-3-hydroxy-5,9,17-trioxoandrosta-1(10),2-diene-4-oic acid (4,9-DHSA) and is involved in cholesterol catabolism (Van der Geize et al., 2007 ▶). The gene encoding HsaD in M. tuberculosis is found in an operon which was predicted (Payton et al., 2001 ▶) and subsequently shown (Anderton et al., 2006 ▶) to consist of genes encoding HsaA, HsaD, HsaC, HsaB (Van der Geize et al., 2007 ▶), a hypothetical protein and an arylamine N-acetyltranferase (NAT), as shown in Fig. 1 ▶. The nat gene has been shown to be required for intracellular survival of the M. tuberculosis model organism M. bovis BCG inside macrophage cells (Bhakta et al., 2004 ▶). The phenotype following ablation of the nat gene in M. bovis BCG was mimicked by growing the wild-type organism in the presence of a putative substrate of HsaC to act as a competitive inhibitor of this enzyme (Anderton et al., 2006 ▶). Large-scale transposon-mutagenesis studies have suggested that the genes encoding HsaA and HsaD are also essential for intracellular survival of M. tuberculosis in human macrophages (Rengarajan et al., 2005 ▶). Recent work has demonstrated that this operon is part of a larger regulon that is involved in lipid metabolism (Kendall et al., 2007 ▶). Therefore, the enzymes encoded by the genes in this operon are important for understanding the biology of M. tuberculosis and also represent potential therapeutic targets.
Figure 1.

The hsa operon in M. tuberculosis and M. bovis BCG. The accession numbers for the genes in M. tuberculosis H37Rv, ordered from right to left, are Rv3570c, Rv3569c, Rv3568c, Rv3567c, Rv3566A and Rv3566c. The corresponding genes in M. bovis BCG have accession numbers Mb3601c, Mb3600c, Mb3599c, Mb3598c, Mb3597c and Mb3596c.
In this paper, we report the 2.35 Å resolution structure of HsaD from M. tuberculosis solved by X-ray crystallography at the Diamond Light Source synchrotron.
2. Materials and methods
2.1. Production and purification of HsaD
The HsaD open reading frame was cloned from M. tuberculosis strain H37Rv into the expression vector pVLT31 with a 20-amino-acid N-terminal hexahistidine tag (amino-acid sequence MGSSHHHHHHSSGLVPR). The expression host used was Pseudomonas putida KT4224. Heterologous expression in LB medium at 303 K gave a typical yield of 15 mg HsaD protein per litre of bacterial culture after purification by immobilized nickel-ion affinity chromatography. The N-terminal hexahistidine tag could not be removed from the recombinant protein by thrombin cleavage. Details of the cloning, expression and protein purification will be published elsewhere.
2.2. Crystallization and structure solution
Purified recombinant HsaD protein (in 100 mM sodium phosphate pH 7.4) was concentrated to 10 mg ml−1 with an Amicon ultracentrifugation concentrator (Millipore, Watford, Hertfordshire). The crystals described in this paper were grown at 292 K by the sitting-drop vapour-diffusion method. For crystallization, 150 nl concentrated HsaD solution was mixed with 150 nl precipitant [30%(w/v) PEG 3000, 0.1 M CHES pH 9.5] and the volume of precipitant in the reservoir was 100 µl. Crystals typically grew within 3–5 d.
Crystals were briefly transferred to a cryoprotectant solution [a 3:1(v:v) mixture of precipitant and glycerol] prior to flash-freezing in liquid nitrogen. Diffraction data were collected from one plate-shaped crystal of approximate dimensions 50 × 50 × 10 µm at beamline IO4 at the Diamond Light Source, Oxfordshire, England. Data from 178 images (oscillation range 0.5°) were indexed and integrated with MOSFLM (Leslie, 1992 ▶) and scaled with SCALA (Evans, 2006 ▶). Initial phases were determined by molecular replacement with the program Phaser (Read, 2001 ▶) by using an ensemble of the MCP hydrolases from Burkholderia xenovorans LB400 (PDB code 2og1; Horsman et al., 2006 ▶) and Rhodococcus jostii RHA11 (PDB code 1c4x; Nandhagopal et al., 1997 ▶) as search templates, with nonconserved residues truncated to the Cβ atom with the program CHAINSAW (Schwarzenbacher et al., 2004 ▶). Initial model building was performed with ARP/wARP (Morris et al., 2003 ▶) and was followed by iterative cycles of manual building with Coot (Emsley & Cowtan, 2004 ▶), refinement with REFMAC (Murshudov et al., 1997 ▶) or phenix.refine (Adams et al., 2002 ▶) and model-quality checking with MOLPROBITY (Davis et al., 2007 ▶). The protein model was solvated and the waters checked with phenix.refine and Coot. The stereochemical quality of the final model was assessed with the programs MOLPROBITY and PROCHECK (Laskowski et al., 1993 ▶). Data-collection and refinement statistics are shown in Table 1 ▶.
Table 1. Summary of data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Space group | I212121 |
| Unit-cell parameters | a = 82.36, b = 82.46, c = 194.67, α = β = γ = 90 |
| Data-collection statistics | |
| Wavelength (Å) | 0.96800 |
| Resolution (Å) | 32.4–2.35 (2.48–2.35) |
| No. of unique reflections | 24657 (3312) |
| Rmerge† | 0.075 (0.50) |
| I/σ(I) | 7.9 (1.5) |
| Completeness (%) | 96.7 (93.6) |
| Redundancy | 3.4 (3.4) |
| Refinement and model statistics | |
| Resolution (Å) | 32.4–2.35 (2.40–2.35) |
| No. reflections used (work + test) | 24657 |
| Rwork‡ | 0.212 (0.305) |
| Rfree‡ | 0.233 (0.294) |
| No. of residues (chain A/chain B) | 284/284 |
| No. of water molecules | 208 |
| Additional molecules | 3 glycerol, 2 sulfate |
| Total No. of atoms | 4624 |
| R.m.s.d. bond lengths (Å) | 0.015 |
| R.m.s.d. bond angles (°) | 0.81 |
| Mean B factor (Å2) | 54.1 |
| Ramachandran statistics (%) | |
| Core region | 88.4 |
| Additional allowed region | 11.2 |
| Generously allowed | 0.4 |
| Disallowed | 0.0 |
R
merge = 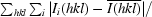
 , where Ii(hkl) is the intensity of the ith observation of unique reflection h.
, where Ii(hkl) is the intensity of the ith observation of unique reflection h.
R
work and R
free = 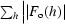 −
− 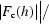
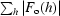 for the working set and test set (5%) of reflections, where F
o(h) and F
c(h) are the observed and calculated structure-factor amplitudes for reflection h.
for the working set and test set (5%) of reflections, where F
o(h) and F
c(h) are the observed and calculated structure-factor amplitudes for reflection h.
3. Results and discussion
The recombinant M. tuberculosis HsaD protein was produced, purified and crystallized as described in §2. A summary of the data-collection and refinement statistics is presented in Table 1 ▶. The asymmetric unit of the M. tuberculosis HsaD crystal structure consists of two chains, each of 284 amino-acid residues (Leu7–Gly290). The electron density was not sufficiently well resolved to model the 26 N-terminal residues, which include the 20-amino-acid hexahistidine affinity tag used for protein purification, or the C-terminal arginine residue.
The M. tuberculosis HsaD enzyme shares only modest amino-acid sequence identity with other MCP hydrolases, including MhpC from Escherichia coli (Dunn et al., 2005 ▶) and the BphD enzymes from B. xenovorans LB400 (Horsman et al., 2006 ▶) and R. jostii RHA1 (Nandhagopal et al., 1997 ▶), as shown in Table 2 ▶. Despite these modest sequence identities, the M. tuberculosis HsaD protein adopts a three-dimensional fold which is highly similar to those of these known MCP hydrolases. The root-mean-square deviations of the Cα backbone atoms between M. tuberculosis HsaD and each of these three MCP hydrolases are given in Table 2 ▶. Fig. 2 ▶ shows a sequence alignment of these four proteins; an overlay of their three-dimensional structures is shown in Fig. 3 ▶.
Table 2. Comparison of the sequence identities and three-dimensional similarity of four MCP hydrolases.
Figure 2.

Sequence alignment of the HsaD protein from M. tuberculosis with BphDs from B. xenovorans (PDB code 2og1) and R. jostii RHA1 (PDB code 1c4x) and MphC from E. coli (PDB code 1u2e). The structure of HsaD from M. tuberculosis was used to generate the structural annotations. The catalytic triad residues Ser114, His269 and Asp241 (M. tuberculosis HsaD numbering) are indicated by black stars. The alignment was performed with ClustalW (Chenna et al., 2003 ▶) and the figure was prepared with ESPRIPT (Gouet et al., 1999 ▶).
Figure 3.

(a, b) Orthogonal views of the ribbon structurs of HsaD from M. tuberculosis (PDB code 2vf2, purple) aligned with those of the BphD enzymes from B. xenovorans LB400 (PDB code 2og1, green) and R. jostii RHA1 (PDB code 1c4x, cyan) and of MphC from E. coli (PDB code 1u2e, yellow). (c) A view of the overlay highlighting the region correlating to amino acids 199–232 in M. tuberculosis HsaD. (d) A view highlighting the α-helical lid domain of M. tuberculosis HsaD. The structural alignments were performed by secondary-structure matching (SSM) in CCP4mg (Krissinel & Henrick, 2004 ▶; Potterton et al., 2004 ▶).
In all four crystal structures shown in Fig. 3 ▶ an α-helical lid domain (α5–η5, amino acids 153–232) is encompassed by an α/β-domain. The α/β-domain is made up of a central β-sheet consisting of three antiparallel strands followed by five parallel strands (from the N-terminus to the C-terminus), surrounded by five α-helices. The active sites of MCP hydrolases are composed of a polar portion (P) and a nonpolar portion (NP) (Seah et al., 2007 ▶), with a well conserved central catalytic triad of residues, Ser114, His269 and Asp241 (M. tuberculosis HsaD numbering). Of these three residues, only the histidine is conserved throughout all known αβ-hydrolases and recent studies have explored the mechanistic role of the active-site histidine residue of BphD from B. xenovorans (Horsman et al., 2007 ▶). The NP subsite of M. tuberculosis HsaD appears to be the entrance to the active-site cleft, in contrast to the proposed active-site entrance (P subsite) of R. jostii RHA1 BphD (Nandhagopal et al., 2001 ▶). One key difference in the three-dimensional fold of M. tuberculosis HsaD relative to these other three MCP hydrolases is the location of amino-acid residues 213–224 (HsaD numbering), which stretches from the C-terminus of α8 to the end of η4 (Fig. 3 ▶) and forms one edge of the NP subsite. The corresponding regions in the other MCP hydrolases are closer to the catalytic triad, resulting in a significantly smaller NP subsite in these proteins relative to M. tuberculosis HsaD (Fig. 4 ▶). The calculated volume of the NP portion of the active-site cavity of M. tuberculosis HsaD (∼2100 Å3) was approximately twofold larger than the corresponding cavity in B. xenovorans BphD (∼1200 Å3) and around fourfold larger than the cavities in the other two MCP hydrolases shown in Fig. 4 ▶ (∼500 Å3), as determined from the PDB files using the program VOIDOO (Kleywegt & Jones, 1994 ▶). This large nonpolar portion of the active site is consistent with the role of M. tuberculosis HsaD in hydrolysis of the cholesterol MCP 4,9-DHSA (Van der Geize et al., 2007 ▶).
Figure 4.

Comparison of the nonpolar (NP) portion of the active-site surfaces of (a) M. tuberculosis HsaD (PDB code 2vf2), (b) BphD from R. jostii RHA1 (PDB code 1c4x), (c) MphC from E. coli (PDB code 1u2e) and (d) BphD from B. xenovorans (PDB code 2og1). The surfaces are coloured by electrostatic potential. The surfaces were calculated for the overlaid structures (Fig. 3 ▶) and the figure was prepared with CCP4mg.
The M. tuberculosis HsaD protein structure was found to adopt a tetrameric assembly in the protein crystal, as shown in Fig. 5 ▶. This tetrahedral structure may be described as a dimer of dimers in which the two monomers in each dimer (green/yellow and blue/magenta in Fig. 5 ▶) are arranged such that an extended 16-strand β-sheet is formed. This tetrahedral assembly was predicted to be stable in solution using the PISA web service at the European Bioinformatics Institute (Krissinel & Henrick, 2007 ▶). The active site of each monomer appears to be distinct and the entrance to each active site (e.g. Fig. 5 ▶ a) is in close proximity to the monomer–monomer β-sheet interface of the opposite dimer (e.g. B–B′). It may therefore be possible for particularly large or long-chain substrates bound at the active site to also interact with residues on the opposite dimer. The equivalent multimeric assemblies of the MCP hydrolases from B. xenovorans (PDB code 2og1) and E. coli (PDB code 1u2e) have also been described (Dunn et al., 2005 ▶; Horsman et al., 2006 ▶).
Figure 5.

The predicted tetrahedral tetrameric assembly of M. tuberculosis HsaD in solution. The molecular assembly was predicted using the Protein Interfaces, Surfaces and Assemblies service (PISA) at the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html; Krissinel & Henrick, 2007 ▶). The assembly may be described as a dimer of dimers in which the monomers coloured green and yellow form one dimer and the monomers coloured blue and magenta form the second dimer. The view in (b) was generated by rotating view (a) by 180° around the y axis. The figure was prepared with CCP4mg.
In summary, the three-dimensional structure of the HsaD protein from M. tuberculosis has been determined by X-ray crystallography at the Diamond Light Source in Oxfordshire, England. The structure shows very high overall similarity to those of other known MCP hydrolases, with the notable exception of one region bounding the active-site pocket. The result of this difference is a larger active site relative to the previously described MCP hydrolases, which is consistent with the substrate specificity of HsaD from M. tuberculosis. The gene encoding HsaD has previously been shown to be essential for the survival of M. tuberculosis inside macrophages and therefore this structure provides a basis for rational ligand design, which will be important both for understanding the biology of M. tuberculosis and for the development of novel antituberculosis agents.
Supplementary Material
PDB reference: HsaD, 2vf2
Acknowledgments
The authors thank the Wellcome Trust for financial support. NL is in receipt of a Canadian National Scholarship (Linacre College, University of Oxford) and a Natural Sciences and Engineering Research Council of Canada (NSERC) Scholarship. LDE acknowledges support from the Canadian Institutes for Health Research (CIHR). The authors also thank Sanjib Bhakta (Birkbeck College, London) for M. tuberculosis genomic DNA and the beamline scientists at IO4 of the Diamond Light Source for technical support.
Footnotes
Formerly Rhodococcus sp. RHA1; the species was recently identified by A. L. Jones and M. Goodfellow (personal communication).
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Anderton, M. C., Bhakta, S., Besra, G. S., Jeavons, P., Eltis, L. D. & Sim, E. (2006). Mol. Microbiol.59, 181–192. [DOI] [PubMed]
- Bhakta, S., Besra, G. S., Upton, A. M., Parish, T., Sholto-Douglas-Vernon, C., Gibson, K. J. C., Knutton, S., Gordon, S., daSilva, R. P., Anderton, M. C. & Sim, E. (2004). J. Exp. Med.199, 1191–1199. [DOI] [PMC free article] [PubMed]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G. & Thompson, J. D. (2003). Nucleic Acids Res.31, 3497–3500. [DOI] [PMC free article] [PubMed]
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B. III, Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids Res.35, W375–W383. [DOI] [PMC free article] [PubMed]
- Dunn, G., Montgomery, M. G., Mohammed, F., Coker, A., Cooper, J. B., Robertson, T., Garcia, J. L., Bugg, T. D. & Wood, S. P. (2005). J. Mol. Biol.346, 253–265. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Horsman, G. P., Bhowmik, S., Seah, S. Y., Kumar, P., Bolin, J. T. & Eltis, L. D. (2007). J. Biol. Chem.282, 19894–19904. [DOI] [PubMed]
- Horsman, G. P., Ke, J., Dai, S., Seah, S. Y., Bolin, J. T. & Eltis, L. D. (2006). Biochemistry, 45, 11071–11086. [DOI] [PMC free article] [PubMed]
- Kendall, S. L., Withers, M., Soffair, C. N., Moreland, N. J., Gurcha, S., Sidders, B., Frita, R., Ten Bokum, A., Besra, G. S., Lott, J. S. & Stoker, N. G. (2007). Mol. Microbiol.65, 684–699. [DOI] [PMC free article] [PubMed]
- Kleywegt, G. J. & Jones, T. A. (1994). Acta Cryst. D50, 178–185. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Morris, R. J., Perrakis, A. & Lamzin, V. S. (2003). Methods Enzymol.374, 229–244. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nandhagopal, N., Senda, T., Hatta, T., Yamada, A., Masai, E., Fukuda, M. & Mitsui, Y. (1997). Proc. Jpn Acad. Ser. B, 73, 154–157.
- Nandhagopal, N., Yamada, A., Hatta, T., Masai, E., Fukuda, M., Mitsui, Y. & Senda, T. (2001). J. Mol. Biol.309, 1139–1151. [DOI] [PubMed]
- Payton, M., Mushtaq, A., Yu, T. W., Wu, L. J., Sinclair, J. & Sim, E. (2001). Microbiology, 147, 1137–1147. [DOI] [PubMed]
- Potterton, L., McNicholas, S., Krissinel, E., Gruber, J., Cowtan, K., Emsley, P., Murshudov, G. N., Cohen, S., Perrakis, A. & Noble, M. (2004). Acta Cryst. D60, 2288–2294. [DOI] [PubMed]
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Rengarajan, J., Bloom, B. R. & Rubin, E. J. (2005). Proc. Natl Acad. Sci. USA, 102, 8327–8332. [DOI] [PMC free article] [PubMed]
- Schwarzenbacher, R., Godzik, A., Grzechnik, S. K. & Jaroszewski, L. (2004). Acta Cryst. D60, 1229–1236. [DOI] [PubMed]
- Seah, S. Y., Ke, J., Denis, G., Horsman, G. P., Fortin, P. D., Whiting, C. J. & Eltis, L. D. (2007). J. Bacteriol.189, 4038–4045. [DOI] [PMC free article] [PubMed]
- Van der Geize, R., Heuser, T., Yam, K., Wilbrink, M., Hara, H., Anderton, M. C., Sim, E., Dijkhuizen, L., Davies, J., Mohn, W. & Eltis, L. D. (2007). Proc. Natl Acad. Sci. USA, 104, 1947–1952. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: HsaD, 2vf2