

Abstract
Dopa decarboxylase (DDC) catalyzes not only the decarboxylation of l-aromatic amino acids but also side reactions including half-transamination of d-aromatic amino acids and oxidative deamination of aromatic amines. The latter reaction produces, in equivalent amounts, an aromatic aldehyde or ketone (depending on the nature of the substrate), and ammonia, accompanied by O2 consumption in a 1 : 2 molar ratio with respect to the products. The kinetic mechanism and the pH dependence of the kinetic parameters have been determined in order to obtain information on the chemical mechanism for this reaction toward 5-hydroxytryptamine (5-HT). The initial velocity studies indicate that 5-HT and O2 bind to the enzyme sequentially, and that d-Dopa is a competitive inhibitor versus 5-HT and a noncompetitive inhibitor versus O2. The results are consistent with a mechanism in which 5-HT binds to DDC before O2. The pH dependency of log V for the oxidative deaminase reaction shows that the enzyme possesses a single ionizing group with a pK value of ∼7.8 that must be unprotonated for catalysis. In addition to an ionizing residue with a pK value of 7.9 similar to that found in the V profile, the (V/K)5-HT profile exhibits a pK value of 9.8, identical to that of free substrate. This pK was therefore tentatively assigned to the α-amino group of 5-HT. No titrable ionizing residue was detected in the (V/K)O2 profile, in the pH range examined. Surprisingly, at pH values lower than 7, where oxidative deamination does not occur to a significant extent, a half-transamination of 5-HT takes place. The rate constant of pyridoxamine 5′-phosphate formation increases below a single pK of ∼6.7. This value mirrors the spectrophotometric pKspec of the shift 420–384 nm of the external aldimine between DDC and 5-HT. Nevertheless, the analysis of the reaction of DDC with 5-HT under anaerobic conditions indicates that only half-transamination occurs with a pH-independent rate constant over the pH range 6–8.5. A model accounting for these data is proposed that provides alternative pathways leading to oxidative deamination or half-transamination.
Keywords: Pyridoxal 5′-phosphate, dopa decarboxylase, serotonin, oxygen, oxidative deamination, half-transamination
Recombinant pig kidney Dopa decarboxylase (DDC; EC 4.1.1.28) is a homodimer that contains 1 mole of pyridoxal 5′-phosphate (PLP) per monomer (Moore et al. 1996). Although DDCs from several sources have been examined, most of the structural and functional information on the enzyme has been obtained from pig kidney (for review, see Moore et al. 1996; this paper) and rat liver (Dominici et al. 1987; Hayashi et al. 1993; Nishino et al. 1997). Besides decarboxylation of l-aromatic amino acids, which is the main reaction, the enzyme catalyzes recently identified side reactions, that is, the oxidative deamination of aromatic amines (Scheme 1 ▶; Bertoldi et al. 1996, 1998), and the half-transamination of d-aromatic amino acids accompanied by a Pictet–Spengler reaction (Bertoldi et al. 1999b). It is also worth noting that, although with d-aromatic amino acids the reaction specificity of DDC does not change in the presence or absence of O2 (Bertoldi and Borri Voltattorni 2000), it appears to be affected by O2 when the enzyme reacts with l-aromatic amino acids or aromatic amines (Bertoldi et al. 1999b; Bertoldi and Borri Voltattorni 2000). In Table 1 the reactions catalyzed by DDC toward l-Dopa, d-5-hydroxytryptophan (5-HTP), and 5-hydroxytryptamine (5-HT, serotonin), as well as their rates under aerobic and anaerobic conditions are reported.
Figure .

Scheme 1. A model for the alternative pathways leading to oxidative deamination or half-transamination of 5-HT by DDC.
Table 1.
Reactions catalyzed by DDC toward l-Dopa, d-5HTP and 5-HT and their relative rates in the presence or absence of O2 in 50 mM Hepes at pH 7.5

kdecarb is the initial velocity expressed as moles of dopamine/min per mole enzyme, kox decarb and khalf trans are the rate constants of PMP formation expressed as min−1, kox deam is the initial velocity expressed as moles of ammonia or 5-hydroxyindolacetaldehyde/min per mole enzyme; n.d. = not detectable.
a Bertoldi and Borri Voltattorni (2000).
b This work.
c Bertoldi et al. (1998).
Oxidative deamination of aromatic amines is the only one of these reactions that does not seem to proceed by any of the known pathways for PLP-catalyzed reactions. It leads to formation, in equivalent amounts, of aromatic aldehyde or ketone (depending on the nature of the substrate), and ammonia accompanied by O2 consumption in a 1 : 2 molar ratio with respect to the products (Bertoldi et al. 1996, 1998). We have also demonstrated that this reaction is operative for other α-decarboxylases, including Lactobacillus 30a ornithine decarboxylase and Escherichia coli glutamate decarboxylase (Bertoldi et al. 1999a). However, the mechanism through which the O2-dependent chemistry occurs remains an intriguing question. In this regard, Abell and Schloss (1991) have shown that several enzymes mediating carbanion chemistry are capable of reacting with molecular oxygen, thus catalyzing oxygen-consuming side reactions. Glutamate decarboxylase, a PLP-enzyme, was one of these enzymes. The following mechanism involving the formation of a peroxide anion and its stabilization either through protonation or through metal coordination was proposed:
As a first step in mechanistic analysis, the kinetic mechanism of the oxidative deamination of DDC on 5-HT has been determined. The results reported here are consistent with ordered addition of 5-HT to the enzyme followed by O2. It is also established that whereas in anaerobiosis DDC only catalyzes a pH-independent half-transamination of 5-HT, in aerobiosis it exhibits a high pH oxidative deaminase activity, and a low pH half-transaminase activity. pH profiles of kinetic parameters for these reactions, along with the pH dependence of the absorbance spectral features of the 5-HT–enzyme intermediate complexes, indicate that two ionizing groups are involved in the catalytic pathways of these reactions. On the basis of these results, hypotheses for the chemical mechanism of these reactions are proposed.
Results
Reaction of DDC with 5-HT in the presence of nitrobluetetrazolium (NBT)
An oxidative deamination occurs when DDC is incubated with 5-HT under aerobic conditions leading to production of 5-hydroxyindolacetaldehyde and ammonia (Scheme 1 ▶; Bertoldi et al. 1996) and O2 consumption in a 1 : 2 molar ratio with respect to the products (Bertoldi et al. 1998). Under anaerobic conditions, this reaction does not occur to a significant extent (Bertoldi et al. 1998). To determine whether molecular oxygen could be replaced by another electron acceptor in the oxidative deamination, 8 μM DDC was incubated in anaerobiosis with 4 mM 5-HT in 50 mM HEPES (pH 7.5) at 25°C, in the presence of 0.1 or 1 mM NBT. No change in absorbance at 560 nm (indicative of reduction of the dye) and no production of aldehyde or ammonia could be observed during the 1-h reaction.
Steady-state kinetics studies of the reaction of DDC with 5-HT
Initial velocity kinetic studies of the oxidative deamination reaction were carried out in 50 mM HEPES (pH 7.5) by varying the 5-HT concentration at several different fixed O2 concentrations. These data yielded linear reciprocal plots (either vs 1/5-HT or 1/O2) that intersected to the left of the Y-axis (data not shown). Although this finding rules out a rapid equilibrium-ordered Bi Bi mechanism, it is diagnostic of a sequential mechanism (Segel 1975). The data were fitted to eq. 1 below and yielded values for K of 5-HT and O2 of 0.25 ± 0.11 and 3.8 ± 0.7 mM, respectively; V (in moles of O2 consumed/mole enzyme per minute) was 12.2 ± 1.9/min. The dissociation constant for 5-HT from the binary complex was 0.14 ± 0.07 mM. Inhibition studies were performed to distinguish a rapid equilibrium random from a steady-state ordered mechanism (Segel 1975). d-Dopa, a substrate analog, was found to be a competitive inhibitor versus 5-HT with Ki = 37.9 ± 0.9 μM, and noncompetitive versus O2 with Ki = 18 ± 4 μM and Kis = 4.58 ± 0.23 mM. Although the products analysis is not complete because no good competitive inhibitors versus O2 are available, our data are consistent with a model of ordered addition of 5-HT to the enzyme followed by molecular oxygen.
Spectral change of DDC with 5-HT as a function of pH
The UV-visible absorbance spectra of the recombinant DDC display two peaks at 335 and 425 nm characteristic of the bound coenzyme. These coenzyme absorbing bands show a modest pH dependence (Moore et al. 1996). Addition of saturating concentrations of 5-HT to DDC at pH 7.5 causes the immediate decrease of the absorbance at 335 nm and the concomitant appearance of an absorbance band at 384 nm (Bertoldi et al. 1996). Spectral changes induced under steady-state conditions by addition of 5-HT to the enzyme were examined over the pH range of 6.3–8.4. Although the maximum for the high pH form is 384 nm, the low pH form of the intermediate complex absorbs maximally at 420 nm (Fig. 1 ▶). According to our previous suggestions (Fiori et al. 1975; Borri Voltattorni et al. 1983), it can be proposed that the 420- and 384-nm absorption bands are caused by 4′-N-protonated and 4′-unprotonated Schiff bases, respectively; therefore, the observed transition 420 → 384 nm should be caused by deprotonation of the 4′-nitrogen of the enzyme–5-HT complex (Scheme 1 ▶). The same spectral transition occurs under anaerobic conditions (data not shown). However, the spectra do not show an isosbestic point, suggesting the presence of multiple ionizations or intermediates. Nevertheless, when we fitted the data of the absorbances at 384 and 440 nm as a function of pH to curves with one, two, and three ionizations, we found that they fit best to a model with one ionization: the pKspec values obtained were 6.73 ± 0.1 and 6.9 ± 0.19 for the absorbance at 384 and 440 nm, respectively (Fig. 1 ▶, inset).
Fig. 1.

pH dependence of the UV-visible spectra of DDC in the presence of 5-HT. Spectra of 5.8 μM DDC plus 50 mM 5-HT run in 50 mM HEPES (pH 6.35, 6.5, 6.6, 6.69, 6.83, 6.89, 7.05, 7.5, 7.6, 8.41). Theoretical fit of enzyme–5HT intermediate complexes according to eqs. 6 and 7: 384 nm (pKspec = 6.73 ± 0.1, filled squares, eq. 6) and 440 nm (pKspec = 6.9 ± 0.19, filled triangles, eq. 7). Spectra were made under aerobic conditions, but the same results were obtained under anaerobic conditions (see text).
To further characterize the enzyme–5-HT intermediate complex, CD spectra were recorded for DDC in the presence of saturating concentrations of 5-HT at three pH values. As shown in Figure 2 ▶, after addition of 5-HT to DDC, the CD spectrum displays, in addition to a positive dichroic signal in the 340-nm region, positive dichroic bands centered at 425, 412, or 384 nm at pH 6.3, 6.85, or 8, respectively.
Fig. 2.

CD spectra of DDC in the presence of 5-HT. CD spectra of 10 μM DDC recorded immediately after the addition of 50 mM 5-HT in 50 mM HEPES, at pH values indicated. Spectra were made under aerobic conditions. At the same pH values, CD spectra of DDC in the absence of 5-HT (displaying positive dichroic bands at 420 and 335 nm) are quite similar to each other.
Reactions catalyzed by DDC on 5-HT as a function of pH
The pH dependence of (V)5-HT and (V/K)5-HT (see Materials and Methods) for the oxidative deamination of 5-HT was determined over the pH range 7–9.2 while varying 5-HT concentration at a fixed subsaturating O2 concentration. Assays at pH values above 9.2 are not reliable because a partial dissociation of coenzyme takes place. The effect of pH on (V)O2 and (V/K)O2 was only studied in the pH range 7–8.5 because at pH values higher than 8.5 and at O2 concentrations higher than the atmospheric oxygen concentration, 5-HT undergoes rapid oxidation. Data for both log(V)5-HT and log(V)O2 fit well using eq. 4 below to a curve with a slope of +1, indicating the dependence on an ionizing group with a pK of 7.87 ± 0.006 or 7.77 ± 0.02, respectively (Fig. 3 ▶). The variation with pH of log(V/K)5-HT yielded a curve with a slope of +2. The data fit well with eq. 5 below to yield pK values of 7.94 ± 0.27 and 9.8 ± 0.6 (Fig. 3 ▶), this latter value being outside of the pH range of the assays. On the contrary, the (V/K)O2 profile is wave shaped with a value differing only by a factor of 2 per pH unit (data not shown).
Fig. 3.

pH dependence of the kinetic parameters of the oxidative deamination for 5-HT. The (V)5-HT (filled circles), (V)O2 (open squares), and (V/K)5-HT (open circles) profiles are from a fit of the resulting values using eqs. 4 and 5, respectively. (filled circles) (V)5-HT values, obtained at varying 5-HT concentrations and at atmospheric O2 concentration, are expressed as moles of ammonia produced/mole enzyme per minute; (open squares) (V)O2 values, obtained at 50 mM 5-HT and at varying O2 concentrations, are expressed as moles of O2 consumed/mole enzyme per minute. The slopes of the plots derived from fits of the data are indicated by dashed lines.
We have previously observed that, in addition to the products of oxidative deamination, a small percentage (∼5%) of the original coenzyme was found as pyridoxamine 5′-phosphate (PMP) after 2 h reaction of DDC with 4 mM 5-HT at pH 7.5. This finding was attributed to transaminating events occurring with a very low frequency with respect to the deaminating ones (Bertoldi et al. 1996). The evaluation of PMP formation during the reaction of DDC with 5-HT was extended over a wide pH range. Surprisingly, we found that at pH values lower than 7, where oxidative deamination does not take place to a significant extent, DDC catalyzes half-transamination of 5-HT as the major reaction. The rate constant (k) of PMP formation was measured at saturating 5-HT concentrations in the pH range of 6.0–7.5, and data were fitted to eq. 8 below. As shown in Figure 4 ▶, the pH profile for log k increases below a single pK of 6.72 ± 0.2. No detectable PLP–l-5-HT Pictet–Spengler adduct was found under aerobic conditions over the pH range 6–8.5.
Fig. 4.

Dependency on pH of the rate constant of PMP form during the reaction of DDC with 5-HT. Plots of the rate constant of PMP formation during the reaction of 8 μM DDC with 50 mM 5-HT under aerobic (filled circles) or anaerobic conditions (filled triangles). The curve in the presence of O2 is from a fit of the data using eq. 4 in which the H and K1 variables were exchanged. The line in the absence of O2 represents the average value of the parameter. The slope of the plot derived from the data is indicated by the dashed line.
When reaction of DDC with 5-HT was carried out under anaerobic conditions, only half-transamination occurs over the pH range 6–8.5. The rate constant of PMP formation is pH-independent with a value of 0.098 ± 0.011 min−1, which is identical to that extrapolated on the acidic side in the presence of O2 (Fig. 4 ▶). After 1-h reaction of DDC with 5-HT in an oxygen-free atmosphere, although PLP–l-5-HT cyclic adduct was not detected in the pH range 6–7, it was found in amounts ranging from 5% to 8% of the original PLP content over the pH range 7–8.5.
The half-transamination reaction of DDC with 5-HT has been characterized in the absence of O2. Under these conditions only this reaction takes place. As described above, addition of 5-HT to the enzyme causes the immediate appearance of the 420-nm or 384-nm absorbance bands, depending on pH. These peaks then decreased with time (data not shown). A description of the spectral events in the region below 350 nm was hampered by the large absorbance of the indolic compound at 280 nm. After 30 min of reaction of DDC with 5-HT at 25°C (pH 7.5) under anaerobic conditions, the reaction mixture was divided in two portions. One of them was reduced with NaBH4 (in order to inactivate the remaining PLP bound before conditions change from anaerobic to aerobic) and transferred to a Centricon −30 tube. After centrifugation at 5000g for 10 min, the filtrate was analyzed by HPLC, and PMP was detected in the filtrate. The other portion was denatured and, after removal of the precipitate by centrifugation, was subjected to HPLC. The analysis indicates that 1.9 nmol of 5-hydroxyindolacetaldehyde (measured as 5-hydroxyindole ethyl alcohol, see Materials and Methods) and 1.5 nmol of PMP were found in the supernatant. Reaction of DDC with 5-HT was allowed to proceed until the original PLP content was converted into 85% PMP and 6% PLP–5-HT cyclic adduct. After removal of the unreacted 5-HT and of the reaction products by gel filtration on a PD-10 column, the enzyme exhibits 10% of residual decarboxylase activity compared to the control enzyme. Exogenous PLP added to this inactive enzyme restored completely (92%) the original activity.
When the apoDDC was incubated with PMP and oxo acids (pyruvic acid, phenylpyruvic acid, or α-ketoglutaric acid), we could not register the formation of PLP.
Discussion
A prerequisite for the occurrence of oxidative deamination of 5-HT by DDC is the presence of molecular oxygen in the reaction mixture. In fact, in the absence of O2 oxidative deamination does not occur to a significant extent. Participation of oxygen as substrate has also been proved by its consumption during the reaction (Bertoldi et al. 1998). Further evidence for the O2-dependent nature of the chemistry of the reaction is the finding that replacement of O2 with an artificial electron acceptor, NBT, does not allow the oxidative deamination to take place.
We have investigated the kinetic mechanism of oxidative deamination of 5-HT catalyzed by DDC. The data yielded linear reciprocal plots that intersected to the left of the Y-axis, and there is no evidence of substrate inhibition by 5-HT. The intersecting reciprocal plots indicate that substrate addition is sequential: the order of substrate addition was determined from inhibition studies. d-Dopa exhibited competitive inhibition versus 5-HT and noncompetitive inhibition versus O2. These data are consistent with, but do not prove, a mechanism in which 5-HT binds to the enzyme before O2. Although the initial velocity kinetic pattern does not support the possibility that O2 binds to the free enzyme in a rapid equilibrium, the possibility that O2 could bind productively to DDC in the absence of 5-HT cannot be excluded.
Neither the role of acid–base catalysts in the oxidative deamination nor the protonation state of 5-HT undergoing this reaction has been previously defined. Thus, the pH dependence of the spectral and kinetic properties of the 5-HT–enzyme intermediate complexes has been examined. The absorption spectral change observed upon addition of 5-HT to DDC over the pH range 6.3–8.4 shows two intermediate complexes absorbing at 420 and 384 nm, which are displayed, either in the presence or absence of O2, by the low and high pH forms of the 5-HT–enzyme complex, respectively. The apparent pKspec of this spectral transition is about 6.8 and could be attributed to the deprotonation of the 4′-nitrogen of the 5-HT–enzyme complex (Scheme 1 ▶). Addition of 5-HT to DDC at pH values below, above, or equal to the pKspec results in distinctly different changes of the dichroic coenzyme bands. This suggests that binding of 5-HT to the enzyme causes different changes in the PLP microenvironment with respect to the neighboring residues, depending on pH.
The course of product formation from oxidative deamination of 5-HT by DDC as a function of pH has been determined in order to correlate it with the observed spectral changes taking place in the bound coenzyme. The pH dependency of both log(V)5-HT and log(V)O2 for the oxidative deaminase reaction indicates that a single ionizing group, not yet identified, with a pK value of ∼7.8 must be unprotonated to achieve maximum velocity. This pK coincides approximately with the pK ∼7.9 observed in the (V/K)5-HT profile. If this assignment is correct, it can be concluded that this group is involved in catalysis but not in binding. However, this pK was not observed in the (V/K)O2 profile. One possible explanation is that the pK is perturbed to a value that is out of the accessible range of the (V/K)O2 profile. The pK could be perturbed by (1) a slow step not included in the rate expression of (V/K), (2) a change of a more hydrophobic environment around the ionizable residues, or (3) a hydrogen bonding of the enzyme residues to the reactants. It is difficult to assess if or which one of these events could perturb the pK beyond the region of pH in which we can work. Nevertheless, it could also be possible that the pK ∼7.9 observed in the (V/K)5-HT profile does not coincide with the pK of (V)5-HT and (V)O2 profiles. In this case, this pK should identify another group involved in binding and/or catalysis. The two pKD values with an average pK of 7.4 already obtained for the pH dependence of the dissociation constants for aromatic amino acid methyl ester analogs–DDC complexes (Moore et al. 1997) do not clarify this point. The (V/K)5-HT profile exhibits a second ionizing group with a pK value of 9.8, indicating that this group must be in the correct protonation state for the substrate to bind. Because the pK value of the α-amino group of 5-HT is 9.8, the found pK value could reflect ionization of the substrate. However, one has to be careful about the accuracy of this pK value because it is outside the range of measurements.
The unexpected finding was that at low pH, where oxidative deamination of 5-HT does not occur to a significant extent, 5-HT undergoes half-transamination. The basis for these results has been elucidated by examining the pH dependence of the rate of this reaction. The log k profile increases below a pK of 6.7, a value that is in good agreement with the independently determined spectrophotometric pKspec value of the external aldimine. Therefore, in the presence of O2 and depending on pH, oxidative deaminase and half transaminase activities of DDC toward 5-HT seem to be almost mutually exclusive. Under the optimal condition for each reaction the rate of half-transamination is about 3% that of oxidative deamination. On the other hand, when reaction of DDC with 5-HT was carried out under anaerobic conditions, only half-transamination occurs in a pH independent manner over the pH range 6–8.5.
On the basis of these results, we can point out that (1) in the presence of O2, whereas the external aldimine absorbing at 420 nm is competent for half-transamination, the external aldimine absorbing at 384 nm is competent for oxidative deamination; and (2) in the absence of O2, both the 420- and 384-nm absorbing species are competent for half-transamination.
From the available information, a plausible chemical mechanism explaining the occurrence of these reactions at the active site of DDC in the presence or absence of O2 can be suggested (Scheme 1 ▶). According to the previously proposed functional model of the active site of DDC (Bertoldi et al. 1999b), the external aldimine with 5-HT could be in a conformation orienting the Cα—H bond perpendicular to the plane of the cofactor system, and therefore productive for half-transamination. This could imply a protonation on the C4′ of the quinonoid formed after the scission of the Cα—H bond. Under anaerobic conditions, either the 420- or the 384-nm external aldimines seem to follow this catalytic pathway. Nevertheless, in the presence of O2, the fate of these absorbing species appears to be different and could be rationalized in terms of different accessibility of their carbanionic intermediates to molecular oxygen. It can be suggested that the enzyme undergoes a conformational change at low pH that would make the carbanion intermediate inaccessible to O2. However, a mechanistic hypothesis is also possible.
Two quinonoids, QH+ and Q, could form after labilization of the Cα—H bond from the protonated 420-nm and unprotonated 384-nm external aldimines, respectively (Scheme 1 ▶). The former is provided by a protonated iminic nitrogen, thus displaying a lesser carbanionic character than the latter. For this reason, it is reasonable to suggest that although the C4′ of QH+ can be more easily protonated than oxygenated, Q can be more susceptible to be attacked by O2 than by H+ at its C4′. This could explain why in aerobiosis the external aldimines absorbing at 420 nm or at 384 nm are catalytically competent for half-transamination or oxidative deamination, respectively. Nevertheless, it cannot be excluded that O2 might react also with QH+. Taking into account that the reaction of O2 with the quinonoid is a radical process, and not a single-step process, if the imine nitrogen is protonated (as in QH+), electron transfer to oxygen will give a semiquinone radical cation and superoxide. If the imine nitrogen is not protonated (as in Q), electron transfer will give the neutral semiquinone and superoxide. Usually a neutral radical is more stable and easier to form than the radical cation. Oxygenase activity has been revealed in a number of enzymes catalyzing reactions involving carbanionic intermediates, and a mechanism based on the capability of these intermediates to react with O2 has been proposed (Abell and Schloss 1991). Following this view (see Scheme 1 ▶), binding of O2 to C4′ of Q could lead to the formation of a peroxide anion (I in Scheme 1 ▶). Once I is formed, in the absence of stabilization, it could regenerate oxygen and the carbanion. Stabilization could be achieved by protonation of the peroxide anion to hydroxyperoxy PLP–5-HT intermediate (II in Scheme 1 ▶) with an appropriate enzymic group. The residue with pK 7.8 could be a good candidate for this purpose. Heterolysis of the O—O bond of II will produce an H2O leaving group, regenerating PLP and producing the 5-HT imine complex (III in Scheme 1 ▶) that undergoes hydrolysis to 5-hydroxyindolacetaldehyde and ammonia. According to this proposal, the mechanism of oxidative deamination would not involve a ternary complex (DDC–5-HT–dioxygen) as a prerequisite for catalysis. It would rather involve hydrogen abstraction from the binary complex DDC–5-HT prior to oxygen binding. Although this mechanism is merely speculative, it is reminiscent of a mechanism model suggested for urate oxidase from soybean root nodules in which a unimolecular step intervenes between the sequential ordered binding of urate and O2 (Kahn and Tipton 1997). Substrate activation in advance of O2 binding could be a common theme in O2-dependent enzymes. It must indeed be pointed out that the formation of an activated oxygen–pterin complex has been proposed to be the rate limiting step in the reaction catalyzed by phenylalanine hydroxylase (Carr et al. 1995). In addition, kinetic studies on lipoxygenase have provided evidence that molecular oxygen enters the lipoxygenase-catalyzed reaction only after the substrate C—H bond is cleaved (Glickman and Klinman 1996).
Finally, it should be noted that these kinetic and spectroscopic studies on the reaction of DDC with 5-HT indicate that the α-amino-group-protonated form of the substrate binds to DDC. A recent analysis of the acid–base chemistry of the reaction of DDC with Dopa by transient and steady-state kinetics revealed two routes for the association of the enzyme with Dopa. Although the α-amino-group-unprotonated form of substrate preferentially binds to the enzyme, DDC seems also to allow the binding of the α-amino-group-protonated form of the substrate (Hayashi et al. 1999).
To shed light on this new aspect of B6 catalysis, further studies using rapid-scanning UV-visible spectroscopy are now underway to investigate the effect of pH on the pre-steady-state kinetics of the reactions catalyzed by DDC toward 5-HT. This may help in the dissection of mechanistic pathways along with the identification of reaction intermediates.
Materials and methods
Chemicals and buffers
5-HT, 5-hydroxyindole ethyl alcohol, pyruvic acid, phenylpyruvic acid, PLP, PMP, bovine liver l-glutamate dehydrogenase, horse liver alcohol dehydrogenase, HEPES, Ches, α-ketoglutarate, and NBT were Sigma products. All other chemicals were of the highest purity available. The liquid chromatography solvents (HPLC grade) were from Labscan (Ireland). Buffers at 50 mM final concentration were used over the following pH range: HEPES, 6.2–8.4, Ches, 8.4–9.2. Buffers were titrated with NaOH. The ionic strength of these buffers remains essentially constant over these pH range (Ellis and Morrison 1982). Overlaps were obtained when buffer was changed so that correction could be made for spurious buffer effects.
Enzyme preparation and assays
Recombinant DDC was purified to homogeneity as described (Bertoldi et al. 1996; Moore et al. 1996). The enzyme concentration was determined by using EM = 1.3 × 105 M−1 cm−1 (Dominici et al. 1993). Apoenzyme was prepared as described (Moore et al. 1996). Decarboxylase activity was measured as described by Sherald et al. (1973), as modified by Charteris and John (1975). 5-HT solutions were prepared fresh daily, and their concentrations were determined spectrophotometrically (E280, 5500 M−1 cm−1). Oxidative deaminase activity of DDC toward 5-HT was measured by two methods. For kinetic studies performed at atmospheric oxygen concentration, enzyme activity was determined by measuring production of ammonia using the coupled system of glutamate dehydrogenase (Bertoldi et al. 1998). A reaction mixture containing DDC (3.5–5 μM) and 5-HT at various concentrations was incubated for 10 min at 25°C in 50 mM HEPES or Ches buffer at the desired pH. The reaction was stopped by treating the mixture for 2 min at 100°C followed by centrifugation and, after adjustment to pH 7.5, the supernatant was added to a solution containing 1400 μg of glutamate dehydrogenase and 300 μM NADH in 50 mM HEPES at pH 7.5. Oxidative deamination activity proceeds linearly over a 15-min period in the pH range examined. For kinetic studies performed at varying O2 concentrations, enzyme activity was determined by measuring oxygen consumption on a Clark oxygen monitor with an Instech Model 203 Dissolved Oxygen Measuring System equipped with a 0.6-mL microchamber. The temperature of the microchamber was maintained at 25°C with a circulating water bath. O2 concentrations were varied by saturating 5-HT solutions with O2/N2 gas mixtures in which the O2 concentrations were 128, 256, 512, 768, 1024, or 1280 μM. The O2 concentration in air-saturated buffer was 258 μM. Reactions were initiated by the addition of enzyme (typically 10 μL), and the analog signal from the O2-electrode was collected using a Duo • 18 data recording system (WPI). The rate of oxygen consumption was determined by a decrease in dissolved O2 in the reaction solution. Rates of reactions were measured at O2 concentrations that varied from 128 to 925 μM. At each O2 concentration, the 5-HT concentration was varied between 0.5 and 50 mM. Whereas the pH dependence of V, (V)O2, and V/K (V/K)O2 for O2 was obtained by varying the levels of O2 at saturating concentration of 5-HT, that of V (V)5-HT, and V/K (V/K)5-HT for 5-HT was obtained as a function of 5-HT concentration in the presence of 258 μM O2. Therefore, (V)5-HT and (V/K)5-HT are apparent values because O2 was not saturating. Owing to technical difficulties it is not possible to use saturating oxygen concentration, that is, higher than about 4 mM. The pH's of the reaction mixtures were determined at the temperature of the assay after the initial rates by using a Radiometer Orion 710A equipped with a microelectrode.
In some experiments, oxidative deaminase activity was also measured by a spectroscopic assay using the coupled system with alcohol dehydrogenase from horse liver as described already (Bertoldi et al. 1996).
HPLC analysis
The PLP, PMP, and PLP–5-HT Pictet–Spengler adduct contents during the reaction of DDC with 5-HT were analyzed with the HPLC method described previously (Bertoldi et al. 1999a). Reaction of DDC with 5-HT under anaerobic conditions was performed using 1-mL Reacti-Vials (Aldrich) as described previously (Bertoldi et al. 1998). The standard curves of peak area as a function of the concentration of coenzyme or coenzyme adduct were prepared using commercially available PLP and PMP or coenzyme adduct obtained by synthesis. The production of 5-hydroxyindolacetaldehyde during the reaction of DDC with 5-HT in the absence of O2 was measured with the HPLC method described previously (Bertoldi et al. 1996). After 30 min reaction the aldehyde produced was converted in situ into the more stable compound 5-hydroxyindole ethyl alcohol by adding alcohol dehydrogenase and NADH. The reaction was stopped by heating for 2 min, and the solution was centrifuged to remove the precipitated protein. The supernatant was run on a reverse phase column, and 5-hydroxyindole ethyl alcohol was quantified with a calibration curve from a standard solution.
Spectral measurements
Absorption spectra were recorded in a Jasco V-550 spectrophotometer. The spectrophotometric pK determination of DDC in the presence of 5-HT was determined in 50 mM HEPES at each pH using 5.8 μM enzyme and 50 mM 5-HT. The enzyme solution was drawn through a 0.2-μm filter to reduce light scattering from the small amount of precipitate. The absorbance and the pH of the solution were measured, and a correction was made to account for dilution. Circular dichroism (CD) measurements were carried out in a Jasco J-710 spectropolarimeter at a protein concentration of 10 μM in the presence of 50 mM 5-HT. Spectra were recorded at a scan speed of 50 nm/min with a band width of 2 nm. The 5-HT concentration is sufficient to maintain the enzyme close to saturation throughout the time required for recording the first spectrum.
Data analysis
V and K values at each pH were obtained from nonlinear regression fitting to the Michaelis–Menten equation. Kinetic data were fitted to eqs. 1–3 using the Scientist MicroMath program and to eqs. 4–8 using the Origin MicroCal software. Initial velocities of the reaction of DDC with 5-HT at varying 5-HT concentrations and at several fixed O2 concentrations were determined from linear regression analysis of the time courses, and the data conforming to a general sequential mechanism were fitted to the following eq. 1.
 |
1 |
where A and B are the substrate concentrations, v is the initial velocity, V is the maximal velocity, Ka and Kb are the Michaelis constants for A and B, respectively, and Keqa is the dissociation constant of A from the EA complex.
Competitive inhibition data were fitted to eq. 2, and noncompetitive inhibition data were fitted to eq. 3:
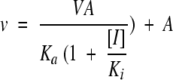 |
2 |
 |
3 |
where [I] is the inhibitor concentration and Ki and Kis are the enzyme-inhibitor or the enzyme substrate complex-inhibitor dissociation constants, respectively.
Data for pH profiles that decreased with a slope of 1 or 2 at low pH were fitted to eqs. 4 and 5, respectively:
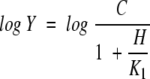 |
4 |
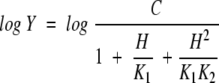 |
5 |
In eqs. 4 and 5, K1 and K2 represent the ionization constants for enzyme or reactant functional groups, Y is the value of the parameter observed as a function of pH, and C is the pH-independent value of Y.
The data recorded at 384 nm and at 440 nm were fitted to eqs. 6 and 7, respectively:
 |
6 |
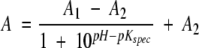 |
7 |
where A1 and A2 are the higher and lower absorbance limits at a particular wavelength, respectively.
The rate constant (k) of formation of PMP was obtained from fitting the PMP concentration-versus-time curve to eq. 8:
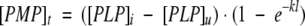 |
8 |
Here, [PMP]t is the measured coenzyme concentration and [PLP]i and [PLP]u are the concentrations of the initial PLP content and of the unreacted coenzyme, respectively. The pH dependence of log k was fitted to eq. 4 except that H and K1 variables are exchanged.
Acknowledgments
This work was supported by funding from the Italian Ministero dell'Università e Ricerca Scientifica e Tecnologica and from CIB (Consorzio Interuniversitario per le Biotecnologie).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
Dopa, 3,4-dihydroxyphenylalanine
DDC, Dopa decarboxylase
PLP, pyridoxal 5′-phosphate
PMP, pyridoxamine 5′-phosphate
5-HTP, 5-hydroxytryptophan
5-HT, 5-hydroxytryptamine
NBT, nitrobluetetrazolium
(V)5-HT, apparent Vmax
(V/K)5-HT, apparent Vmax/K5-HT. See Materials and Methods for information relevant to values
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.46601.
References
- Abell, L.M. and Schloss, J.V. 1991. Oxygenase side reactions of acetolactate synthase and other carbanion-forming enzymes. Biochemistry 30 7883–7887. [DOI] [PubMed] [Google Scholar]
- Bertoldi, M. and Borri Voltattorni, C. 2000. Reaction of dopa decarboxylase with l-aromatic amino acids under aerobic and anaerobic conditions. Biochem. J. 352 533–538. [PMC free article] [PubMed] [Google Scholar]
- Bertoldi, M., Moore, P.S., Maras, B., Dominici, P., and Borri Voltattorni, C. 1996. Mechanism-based inactivation of Dopa decarboxylase by serotonin. J. Biol. Chem. 271 23954–23959. [DOI] [PubMed] [Google Scholar]
- Bertoldi, M., Dominici, P., Moore, P.S., Maras, B., and Borri Voltattorni, C. 1998. Reaction of Dopa decarboxylase with α-methylDopa leads to an oxidative deamination producing 3,4-dihydroxyphenylacetone, an active site directed affinity label. Biochemistry 37 6552–6561. [DOI] [PubMed] [Google Scholar]
- Bertoldi, M., Carbone, V., and Borri Voltattorni, C. 1999a. Ornithine and glutamate decarboxylases catalyse an oxidative deamination of their α-methylsubstrates. Biochem. J. 342 509–512. [PMC free article] [PubMed] [Google Scholar]
- Bertoldi, M., Frigeri, P., Paci, M., and Borri Voltattorni, C. 1999b. Reaction specificity of native and nicked 3,4-dihydroxyphenylalanine decarboxylase. J. Biol. Chem. 274 5514–5521. [DOI] [PubMed] [Google Scholar]
- Borri Voltattorni, C., Minelli, A., and Dominici, P. 1983. Interaction of aromatic amino acids in d and l forms with 3,4-dihydroxyphenylalanine decarboxylase from pig kidney. Biochemistry 22 2249–2254 [DOI] [PubMed] [Google Scholar]
- Carr, R.T., Balasubramanian, S., Hawkins, P.C.D., and Benkovic, S.J. 1995. Mechanism of metal-independent hydroxylation by Chromobacterium violaceum phenylalanine hydroxylase. Biochemistry 34 7525–7532. [DOI] [PubMed] [Google Scholar]
- Charteris, A. and John, R.A. 1975. An investigation of the assay of dopamine using trinitrobenzenesulphonic acid. Anal. Biochem. 66 365–371. [DOI] [PubMed] [Google Scholar]
- Dominici, P., Tancini, B., Barra, D., and Borri Voltattorni, C. 1987. Purification and characterization of rat liver 3,4-dihydroxyphenylalanine decarboxylase. Eur. J. Biochem. 169 209–213. [DOI] [PubMed] [Google Scholar]
- Dominici, P., Moore, P.S., and Borri Voltattorni, C. 1993. Dissociation, unfolding and refolding trials of pig kidney 3,4-dihydroxyphenylalanine (Dopa) decarboxylase. Biochem. J. 295 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, K.J. and Morrison, J.F. 1982. Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 87 405–426. [DOI] [PubMed] [Google Scholar]
- Fiori, A., Turano, C., Borri Voltattorni, C., Minelli, A., and Codini, M. 1975. Interaction of l-Dopa decarboxylase with substrates: A spectrophotometric study. FEBS Lett. 54 122–125. [DOI] [PubMed] [Google Scholar]
- Glickman, M.H. and Klinman, J.P. 1996. Nature of rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry 35 12882–12892. [DOI] [PubMed] [Google Scholar]
- Hayashi, H., Mizuguchi, H., and Kagamiyama, H. 1993. Rat liver aromatic l-amino acid decarboxylase: spectroscopic and kinetic analysis of the coenzyme and reaction intermediates. Biochemistry 32 812–818. [DOI] [PubMed] [Google Scholar]
- Hayashi, H., Tsukiyama, F., Ishii, S., Mizuguchi, H., and Kagamiyama, H. 1999. Acid–base chemistry of the reaction of aromatic l-amino acid decarboxylase and Dopa analyzed by transient and steady-state kinetics: Preferential binding of the substrate with its amino group unprotonated. Biochemistry 38 15615–15622. [DOI] [PubMed] [Google Scholar]
- Kahn, K. and Tipton, P.A. 1997. Kinetic mechanism and cofactor content of soybean root nodule urate oxidase. Biochemistry 36 4731–4738. [DOI] [PubMed] [Google Scholar]
- Moore, P.S., Dominici, P., and Borri Voltattorni, C. 1996. Cloning and expression of pig kidney dopa decarboxylase: Comparison of the naturally occurring and recombinant enzymes. Biochem. J. 315 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, P.S., Bertoldi, M., Dominici, P., and Borri Voltattorni, C. 1997. Aromatic amino acid methyl ester analogs form quinonoidal species with Dopa decarboxylase. FEBS Lett. 412 245–248. [DOI] [PubMed] [Google Scholar]
- Nishino, J., Hayashi, H., Ishii, S., and Kagamiyama, H. 1997. An anomalous side reaction of the Lys303 mutant aromatic l-amino acid decarboxylase unravels the role of the residues in catalysis. J Biochem. (Tokyo) 121 604–611. [DOI] [PubMed] [Google Scholar]
- Segel, I.H. 1975. Enzyme kinetics. Wiley, New York.
- Sherald, A.F., Sparrow, J.C., and Wright, T.R.F. 1973. A spectrophotometric assay for Drosophila Dopa decarboxylase. Anal. Biochem. 56 300–305. [DOI] [PubMed] [Google Scholar]