

Abstract
Shikimate kinase, despite low sequence identity, has been shown to be structurally a member of the nucleoside monophosphate (NMP) kinase family, which includes adenylate kinase. In this paper we have explored the roles of residues in the P-loop of shikimate kinase, which forms the binding site for nucleotides and is one of the most conserved structural features in proteins. In common with many members of the P-loop family, shikimate kinase contains a cysteine residue 2 amino acids upstream of the essential lysine residue; the side chains of these residues are shown to form an ion pair. The C13S mutant of shikimate kinase was found to be enzymatically active, whereas the K15M mutant was inactive. However, the latter mutant had both increased thermostability and affinity for ATP when compared to the wild-type enzyme. The structure of the K15M mutant protein has been determined at 1.8 Å, and shows that the organization of the P-loop and flanking regions is heavily disturbed. This indicates that, besides its role in catalysis, the P-loop lysine also has an important structural role. The structure of the K15M mutant also reveals that the formation of an additional arginine/aspartate ion pair is the most likely reason for its increased thermostability. From studies of ligand binding it appears that, like adenylate kinase, shikimate kinase binds substrates randomly and in a synergistic fashion, indicating that the two enzymes have similar catalytic mechanisms.
Keywords: Shikimate kinase, P-loop lysine, ion pair, enzyme stability
The shikimate pathway is essential to plants and microorganisms for the biosynthesis of aromatic compounds but absent from animals. This pathway is therefore a promising target for the development of potentially nontoxic antimicrobial agents (Davies et al. 1994) and herbicides (Coggins 1989).
Shikimate kinase (SK; EC 2.7.1.71) catalyzes the fifth reaction in this 7-step metabolic route, converting shikimate into shikimate 3-phosphate, using ATP as co-substrate. In Escherichia coli, this reaction is catalyzed by the two isoforms SK I (encoded by the aroK gene; Whipp and Pittard 1995) and SK II (encoded by the aroL gene; Millar et al. 1986). The SK II isoform appears to play a dominant role in the shikimate pathway; its expression is controlled by the tyrR regulator, and it is repressed by tyrosine and tryptophan (Ely and Pittard 1979; De Feyter et al. 1986). The role of SK I appears to be less clear; it is expressed constitutively and has a very much lower affinity for shikimate (Km of 20 mM compared with 200 μM for SK II) (De Feyter and Pittard 1986). An alternative physiological role that is associated with sensitivity to the antibiotic mecillinam (Vinella et al. 1996) has been reported for SK I. However, complete genome sequences of a number of bacteria, for example, Haemophilus influenzae and Mycobacterium tuberculosis, have revealed the presence of only the aroK-encoded SK I enzyme but no aroL-encoded SK II.
SKs contain the consensus sequence GXXXXGKT/S (with X being any residue; Walker et al. 1982), which forms the so-called P-loop, an area of the ATP-binding site that interacts directly with the phosphate groups of bound nucleotides. In addition to a rather strict conservation of this sequence in many ATP- or GTP-binding proteins, the same structural organization of this region is retained across many different families of proteins. The superimposition of the P-loops from proteins such as recA, adenylate kinase (AK), the elongation factors, G-proteins, and myosin result in r.m.s. deviations in Cα atoms of only 0.3–0.4 Å, which can be entirely attributed to coordinate errors (Smith and Rayment 1996). This close similarity of P-loops is remarkable considering the different functions of these proteins. The side chain of the P-loop lysine has been shown to be oriented toward the phosphate groups of bound nucleotides (Müller and Schulz 1988; Pai et al. 1989). It is thought to have a catalytic role of stabilizing the pentavalent transition state of the γ-phosphoryl group as has been documented for p21ras (Sigal et al. 1986) and AK (Reinstein et al. 1990).
The P-loop lysine has been studied extensively by site-directed mutagenesis, but the resulting biochemical parameters of these mutant proteins differ enormously. Kinetic analysis of the mutant proteins often indicates a complete loss in ATP-dependent activity for both conservative and non-conservative exchanges (Deyrup et al. 1998; Hishida et al. 1999). Other reports show that substantial activity is retained when the replacements are conservative (Delepelaire 1994; Kang et al. 1997). For example, the kinetic parameters of the P-loop K16Q mutant of adenylosuccinate synthetase are similar to those of the wild type. The corresponding mutant in AK (K13Q) shows significantly lower activity, with only a modest effect on substrate binding (Reinstein et al. 1990), whereas an analogous mutation in p21ras (K16N) drastically reduces the affinity for nucleotides (Sigal et al. 1986). This wide spectrum of results is astonishing considering the almost complete structural conservation of the P-loop and suggests that, besides being a catalytic residue, the P-loop lysine may also have an important structural role.
We have recently overexpressed, crystallized, and solved the 3d structure of an SK from the plant pathogen Erwinia chrysanthemi (Krell et al. 1997, 1998b). This monomeric enzyme shows 53% amino-acid identity to E. coli SK II and, at less than 19 kD, is one of the smallest kinases so far described. Its small size makes it ideal for structural and mechanistic studies of phosphotransfer. The structure of SK was found to be similar to AK despite a sequence identity of ∼20%. AK has a rapid equilibrium random sequential mechanism (Rhoads and Lowenstein 1968; Font and Gautheron 1980), and undergoes large structural movements upon substrate binding (Schulz et al. 1990). There is also crystallographic evidence of induced-fit movements when SK binds shikimate and ADP (Krell et al. 1998b).
In this paper we demonstrate that the structural similarities between SKs and AKs are also reflected in biochemical and mechanistic similarities. We also report the first structure of a nucleotide-binding protein with a P-loop lysine mutant, SK-K15M, which provides a detailed view of the structural changes that occur as a consequence of this mutation.
Results and Discussion
Characterization of shikimate kinase from E. chrysanthemi
Shikimate kinase (SK) from Erwinia chrysanthemi was predicted to be an SK II-type enzyme on the basis of sequence similarity. The major difference between SK I and SK II of Escherichia coli lies in their affinities for shikimate (Table 1). The values of Km and kcat/Km for the E. chrysanthemi enzyme of 310 μM and 1.3 × 105 M−1 sec−1 confirm this sequence-based prediction.
Table 1.
The kinetic parameters of native and mutant shikimate kinase from Erwinia chrysanthemi in comparison to the Escherichia coli isoenzymes I and II
| Km or Kdc (shikimate) (μM) | Km or Kdc (ATP) (μM) | Kd (ADP) (μM) | kcat (s−1) | kcat/Km (shikimate) (M−1 sec−1) | |
| E. coli | |||||
| (SK I) | 20,000a | — | — | — | |
| (SK II) | 200a | 160a | 27.1b | 1.36 × 105 | |
| E. chrysanthemi | |||||
| Wild type | 310/700c | 620/2560c | 1680 | 35 | 1.3 × 105 |
| C162S | 280 | 670 | 40 | 1.6 × 105 | |
| C13S | 75 | 235/1530c | 1450 | 23 | 3.4 × 105 |
| K15M | 670c | 620c | 290 | n.d.d | n.d. |
| D34N | n.d. | n.d. | n.d.d | n.d. |
Km values were calculated using nonlinear regression fitting to the Michaelis–Menten equation. The assay mix to measure Km (shikimate) contained 5 mM ATP and 3 mM shikimate for Km (ATP).
a Values taken from De Feyter and Pittard (1986).
b Averaged value taken from De Feyter and Pittard (1986) and Millar et al. (1986).
cKd values were determined using fluorescence quenching experiments as described in the Materials and Methods section. These values are dissociation constants of one ligand in the absence of other ligands.
d n.d., no detectable activity or binding.
Binding of substrates to shikimate kinase
The single tryptophan residue (W54) of SK is positioned close to the shikimate binding site and serves as a reporter group to determine the effect of substrate binding using fluorescence spectroscopy. The binding of shikimate leads to a substantial quenching of the fluorescence of SK; the data obtained could be fitted very satisfactorily to a hyperbolic binding model (data not shown). The dissociation constant in the absence of nucleotide was 700 μM; this was reduced in the presence of 1.5 mM ADP to 320 μM, which is comparable to the Km value obtained from steady-state kinetics (see Table 1). The data fitted very well to a hyperbolic binding model (data not shown). The twofold increase in apparent affinity of the enzyme toward shikimate in the presence of nucleotide is strongly supportive of the existence of synergism in substrate binding.
The binding of nucleotides to SK was also measured using fluorescence quenching. The dissociation constants for ADP and ATP were determined to be 1.7 and 2.6 mM, respectively (Table 1). We have also used fluorescence quenching to investigate whether the presence of shikimate alters the affinities for nucleotides. However, the addition of shikimate to SK caused almost complete fluorescence quenching, which made it impossible to quantify the small fluorescence changes caused by the subsequent addition of nucleotide. The Km value for ATP was found to be around two times lower than the Kd (in the absence of shikimate), which is consistent with an increase in affinity for nucleotides in the presence of shikimate (Table 1). These binding and kinetic data indicate that SK behaves in an analogous fashion to the AKs in forming the catalytically active ternary complex in a random fashion. Presumably the conformational changes in the enzyme associated with binding of the first substrate lead to an increase in the affinity for the second substrate.
Site-directed mutagenesis and characterization of mutant enzymes
Within the group of P-loop proteins a number of subclasses can be distinguished, each containing an additionally conserved residue within the Walker type A motif (GXXXXGKT/S; Walker et al. 1982). One subclass is characterized by a conserved cysteine positioned just before the GKS/T sequence. The SK II enzymes belong to this subclass, whereas the SK I enzymes have mainly serine or alanine residues at this position. Interestingly, this cysteine is also found frequently in ATP-binding subunits/domains of ATP transporters such as P-glycoprotein and the maltose transporter MalK (Hyde et al. 1990). To investigate the role of this cysteine we designed the mutant C13S as well as the classic P-loop mutant K15M. Outside of the P-loop a third mutant D34N was prepared because we predicted it to be functionally important. The fourth mutant C162S was prepared because it was presumed to be the cause of significant nonspecific aggregation, and in the native SK crystal structure formed a disulfide bond with C162 from a neighboring molecule (Krell et al. 1998b).
Three of the four purified mutant enzymes were found to be stable under standard conditions. Purified D34N precipitated in standard buffer systems, indicating that D34 may play an important role in maintaining the conformation of the enzyme. This mutant protein was also found to be enzymatically inactive and was not characterized in further detail. The far-UV and near-UV CD spectra of the remaining three mutants and the native SK are very similar, showing that the overall secondary and tertiary structural features of the wild-type enzyme are retained in the C13S, C162S, and K15M mutants. The detailed structural changes that occurred in the K15M mutant are discussed below in the section dealing with the X-ray structure of this protein.
Table 1 summarizes the kinetic and binding parameters of mutant enzymes. The D34N and K15M mutants had no detectable enzyme activity (less than 0.1% of that of the wild-type enzyme). Fluorescence quenching experiments were carried out to see whether the K15M mutant is able to bind substrates. Interestingly, the K15M mutant appeared to bind ATP around fourfold tighter than the wild-type enzyme, whereas the affinity for shikimate was unchanged (Table 1). This was an unexpected result because it is, to our knowledge, the first P-loop lysine mutant that has a substantially increased affinity for ATP. In AK, the Km values (which are often considered to reflect affinities) for ATP of mutant enzyme with the lysine replaced by glutamine, arginine, and alanine were found to be 5–20-fold decreased, whereas the value for the methionine mutant was almost the same as for the wild-type enzyme (Reinstein et al. 1990; Tian et al. 1990; Byeon et al. 1995). The Kd value for ADP binding to the K15M mutant of SK, measured by fluorescence quenching, was about half the value for ATP. For the wild-type and C13S mutants the values for ATP and ADP binding were more similar to each other.
The C13S mutation had only a slightly reduced kcat value compared to the wild-type enzyme (Table 1). This result is analogous to those obtained in site-directed mutagenesis studies of the MalK subunit of the maltose ABC transporter (Hunke and Schneider 1999) and the A subunit of the yeast vacuolar ATPase (Taiz et al. 1994; Liu et al. 1997). In MalK, this cysteine is highly conserved. However, in all three cases the replacement of the cysteine, positioned as in SK before the GKT/S sequence, resulted in fully functional protein, and the catalytic activity or phenotype was indistinguishable from the wild-type protein (Hunke and Schneider 1999). The analysis of the steady-state kinetics and fluorescence quenching measurements for the C13S mutant of SK revealed that substrates bind with a higher affinity to the protein. which resulted in an increase in the catalytic efficiency (kcat/Km) as compared to the wild-type protein (Table 1). Unfortunately, the affinity of the mutant enzymes for substrates has not been investigated in the above-mentioned studies of MalK and vacuolar ATPase.
The catalytic and substrate binding parameters of the C162S mutant were very similar to those of the wild-type protein, which was expected (Table 1). A substantial increase in enzyme solubility was noted, confirming that this residue plays a role in the aggregation of the protein at high concentration, presumably by forming disulphide-linked dimers with either C162 or C13 of another molecule of enzyme.
Probing the reactivity of K15 and C13 using chemical modification with TNBS and 2PDS
The lysine residue K15, located in the P-loop of SK from E. chrysanthemi is the only lysine residue in the enzyme. This makes it possible to study the reactivity of this important residue using TNBS, which is reported to be a lysine-selective reagent (Means and Feeney 1971) (but see below). The reaction of TNBS with amines is strongly pH-dependent as only unprotonated amino groups react (Goldfarb 1966).
Incubation of SK with TNBS results in an extremely rapid decrease in enzyme activity, with 65% activity lost after 4 min reaction with 2.5 μM TNBS. The inactivation obeys pseudo-first-order kinetics and the second-order rate constant k, calculated from the slope of a plot of kobs versus [TNBS] is 6.15 × 104 M−1 min−1. The presence of 1 mM ATP in the inactivation mixture affords substantial (>60%) protection from inactivation, whereas the presence of 1 mM shikimate resulted in no significant change (<10%) in the rate of inactivation. Evidence from mass spectrometry (data not shown) indicates that there was a single site of modification, and together with the protection data, suggests that the inactivation is due to the reaction of K15. Another enzyme of the shikimate pathway, shikimate dehydrogenase, has been shown to have a single lysine residue, which is essential for enzyme activity (Krell et al. 1998a). Under the same conditions as used in this work, the second-order rate constant for the reaction of shikimate dehydrogenase with TNBS was found to be only 400 M−1 min−1, a value comparable with those obtained in studies of the reaction of TNBS with a number of other non-ATP utilizing proteins (Goldin and Frieden 1971; Suzuki et al. 1995).
It is noteworthy that the abnormally high reactivity of wild-type SK toward TNBS is not observed with the C13S mutant, for which k for the inactivation is only 150 M−1 min−1, that is, 400 times lower than the value for the reaction of SK itself. As is found for the reaction of SK, reaction of the mutant with TNBS was slower in the presence of 2 mM ATP, which supports the interpretation that reaction is at the active-center K15 side chain. A possible explanation for the abnormally high reactivity of native SK toward TNBS, not observed with the C13S mutant, involves initial reaction of C13 followed by intramolecular transfer of the trinitrophenyl group to K15. The crystal structure of SK (Krell et al. 1998b) revealed the proximity of these residues, and kinetic studies using 2PDS as a thiol-specific, two-protonic-state reactivity probe (Brocklehurst 1982) discussed below, demonstrate the nucleophilic character of C13 at low pH, diagnostic of a (K15)-N+H3/(C13)-S− ion pair. Although TNBS is commonly considered to be a lysine-specific reagent, it does react also with thiol groups (Freedman and Radda 1968), and S to N transfer analogous to that now suggested for K15 → C13 in SK has been demonstrated in reactions of proteins with 4-chloro-7-nitrobenzofurazan (Birkett et al. 1970).
Reactivity in the thiolate-imidazolium ion pairs of cysteine proteinases (Brocklehurst et al. 1998) has been demonstrated by using 2-pyridyl disulfides such as 2PDS (Plou et al. 1996; Pinitglang et al. 1997). Protonation of the pyridyl N atom as in 2PDSH+ (pKa 2.45) enhances the reactivity of the reagent by ∼1000×. The coexistence of 2PDSH+ and a thiolate anion from a low pKa thiol group (typically pKa 2–4 in ion pairs) results in a rate maximum in acidic media, where unperturbed thiol groups exist as undissociated SH and have negligible reactivity. The pH-k profile for the reaction of native SK is shown in Figure 1a ▶. The data fit well to a simple bell-shaped curve characterized by pKa 2.45 (the pKa value of 2PDSH+) and a pKa value ∼3.9, which is assigned to the thiol group of C13 in proximity to the —N+H3 group of K15. The rate maximum in Figure 1a ▶ necessarily represents reaction in a minor ionization state (Brocklehurst 1994Brocklehurst 1996), because chemical logic requires the reaction to involve —S−/-Im+H (produced by increase in pH across pKa 3.9) and 2PDSH+ (produced by decrease in pH across pKa 2.45) rather than —SH/-Im+H and 2PDS. The other thiol group in SK, that of C162, cannot be the site of reaction with 2PDS because reaction, and indeed the rate maximum at ∼pH 3, persists in the pH- k profile for the reaction of the C162S mutant (Fig. 1b ▶).
Fig. 1.
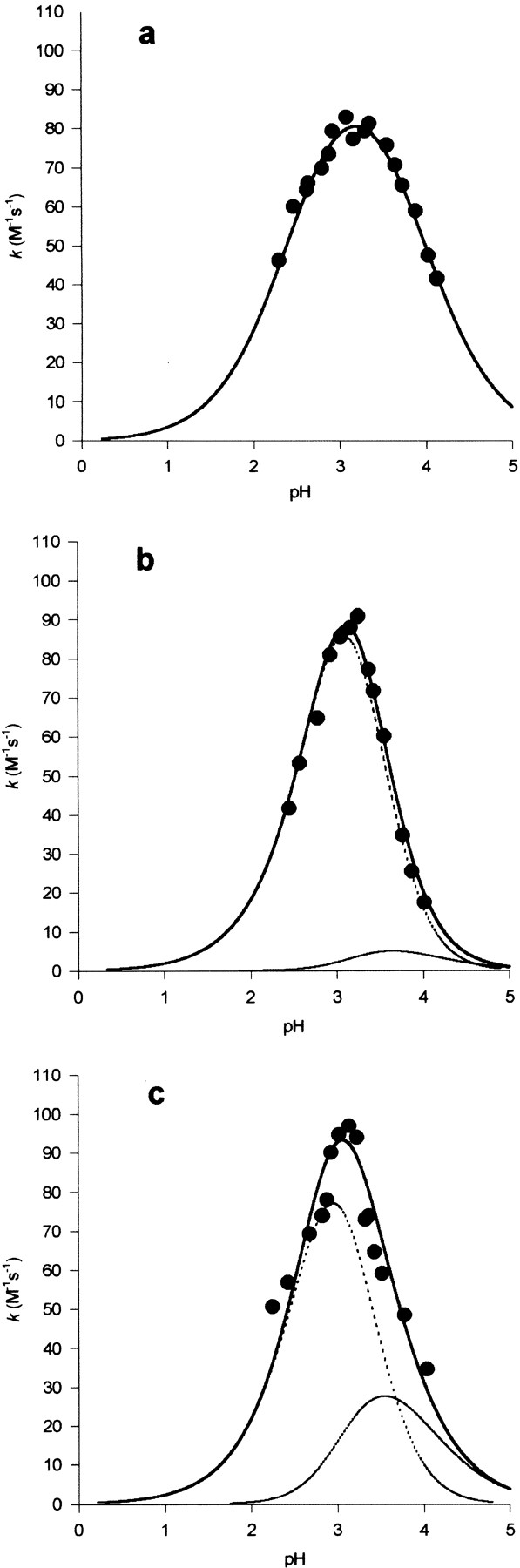
pH-dependence of the second-order rate constant (k) for the reactions at 25°C and I 0.1 M in aqueous buffers containing 1 mM EDTA and 2% (v/v) glycerol of 2PDS with (a) wild-type SK, (b) the C162S mutant, and (c) the K15M mutant. The points are each the mean of 3 determinations (SD ± ≤10% the mean). The continuous lines are theoretical for pH-dependent rate equations in a for 1 reactive protonic state and in b and c for 2 reactive protonic states and the following values of the characterizing parameters (pKa values and pH-independent rate constants, k̃, with values of the latter in M−1 sec−1): (a) pKI = 2.45, pKII = 3.9, k̃= 110; (b) pKI = 3.1, pKII = 3.2, pKIII = 3.9, k̃1 = 250, k̃2 = 10; (c) pKI = 3.0, pKII = 3.0, pKIII = 3.9, k̃1 = 240, k̃2 = 50. In b and c the broken lines are theoretical for the components of the pH-dependent rate equation corresponding to terms in k̃1 and k̃2. In a the broken line is coincident with the continuous line.
The fact that a rate maximum persists in the K15M mutant (Fig. 1c ▶) requires that in this mutant the thiolate anion of C13 is stabilized by something other than (K15)-N+H3. The crystal structure of the K15M mutant described below provides an explanation for this unexpected kinetic result. Thus conformational changes within the P-loop result in displacement of the side chain of C13 out of this motif such that it forms an ion pair with the guanidinium cation of R110.
It is noteworthy that the pH-k profiles in Figure 1 ▶, b and c, are narrower than that in Figure 1a ▶, and that in each case two closely similar pKa values of ∼3 are required to fit the data. The existence of pKa values closer than 0.6 requires cooperative proton binding (Dixon 1973) and suggests that the mutants are susceptible to unfolding in acidic media, resulting in loss of the ion-pair geometry and thus of the existence of the thiolate anion necessary for reaction with 2PDSH+. As a result, reactivity is lost with decrease in pH before the pKa of 2PDSH+ becomes kinetically significant.
The results from the use of TNBS and 2PDS compel the view that the side chains of C13 and K15 form an –S−/ –N+H3 ion pair. The proximity of these side chains provides the C13 thiol group with an abnormally low pKa value of 3.9 and accounts for the apparently abnormally high reactivity of K15 toward TNBS at pH 9.2. Thus high reactivity is achieved by initial reaction of C13 with subsequent intramolecular transfer to K15 to provide the thermodynamically more stable product. The low pKa character of C13 is maintained in the K15M mutant as predicted from the crystal structure, described below, where the side chain of C13 moves to form an interaction with R110.
The functional relevance of this ion pair is not clear, and it would be most interesting to know whether this special reactivity is also observed in other proteins that also have a conserved cysteine in the P-loop. As mentioned above, this cysteine is conserved in many proteins, but its mutation results in fully functional protein (Taiz et al. 1994; Liu et al. 1997; Hunke and Schneider 1999), as is also the case for the SK mutant C13S (Table 1).
It seems possible that the electrostatic interaction of K15 with C13 in the wild-type enzyme contributes to the positioning of the former as a hydrogen-bond donor to the backbone carbonyl oxygens of G9 and T77. These hydrogen bonds are important (see below) for maintaining the conformation of the P-loop.
Differential scanning calorimetry
DSC was used to investigate the effects of mutation and substrate/ligand binding on the thermal stability of SK under near-physiological conditions. As illustrated by protein engineering experiments on yeast AK (Abele and Schulz 1995; Spuergin et al. 1995), which is structurally very similar to SK, the factors influencing protein stability are not well understood. In this study, six site-directed mutant proteins were designed in order to increase the thermal stability of the protein by attempting to introduce structural changes that result in helix capping or core perturbation. However, all mutant proteins were shown to be less stable than the native enzyme, with melting temperatures lowered by 0.6°–7.7°C.
Typical DSC data for SK in the presence and absence of ligands are shown in Figure 2 ▶, and relevant thermodynamic data are collected in Table 2. In all cases the DSC thermograms were characterized by an endothermic transition typical of the heat uptake expected for cooperative unfolding of the protein (Cooper 1999), with some distortion of the thermogram on the high temperature side because of irreversible aggregation of the unfolded protein. Re-scan of such samples showed no further transition. Under such circumstances it is not possible to obtain absolute thermodynamic parameters for the unfolding transition, but the data listed in Table 2 are of qualitative interest. The transition temperature of SK (39°C) was found to be substantially lower than that of E. coli AK (55°C). The C13S mutant was shown to have very similar thermodynamic behavior in comparison to the native enzyme. In contrast, the K15M protein was found to have an increased thermal stability, as expressed by an increase in Tm by 4.2°C. In a similar study by Reinstein et al. (1990), unfolding transition temperatures for the AK P-loop P9G, P9L, and G10V were decreased by ∼6°C, whereas mutant K13Q was found to be only 3°C less stable as compared to the wild-type AK. Inspection of the X-ray structure of the SK mutant reveals that the formation of an ion pair between R11 and D32 (Fig. 5c ▶), which is not pres-ent in the native enzyme, might be the cause for this increase in stability.
Fig. 2.

Raw DSC data illustrating the heat uptake associated with thermal unfolding of shikimate kinase (1 mg/mL) in the presence and absence of substrate molecules (shikimate, ADP, and ATP were at 2 mM). The increase in Tm in the presence of each substrate is consistent with specific ligand binding to the native state of the enzyme.
Table 2.
Effects of ligand binding and site-directed mutations on the thermal stability of shikimate kinase as measured using differential scanning calorimetry
| Ligands | Tm (°C) | ΔTm (°C) | ΔHcal (kJ/mole) | |
| Native SK | — | 39.7 | — | 67 |
| 2 mM shikimate | 41.8 | 2.1 | 80 | |
| 2 mM ADP | 47.3 | 7.6 | 90 | |
| 1 mM ATP | 48.1 | 8.4 | 100 | |
| 2 mM ATP | 49.9 | 10.2 | 98 | |
| 2 mM ADP and shikimate | 50.2 | 10.5 | 69 | |
| C13S | — | 39.0 | −0.7 | 76 |
| 1 mM ATP | 46.1 | 6.4 | 99 | |
| K15M | — | 43.9 | 4.2 | 47 |
| 1 mM ATP | 45.0 | 5.3 | 51 |
Tm is the apparent midpoint of the protein unfolding transition, and ΔHcal is the change in enthalpy on unfolding. ΔTm is the change in transition temperature with respect to native SK in the absence of ligand. Standard errors: ±0.1°C (Tm); ±10% (ΔHcal)
Fig. 5.


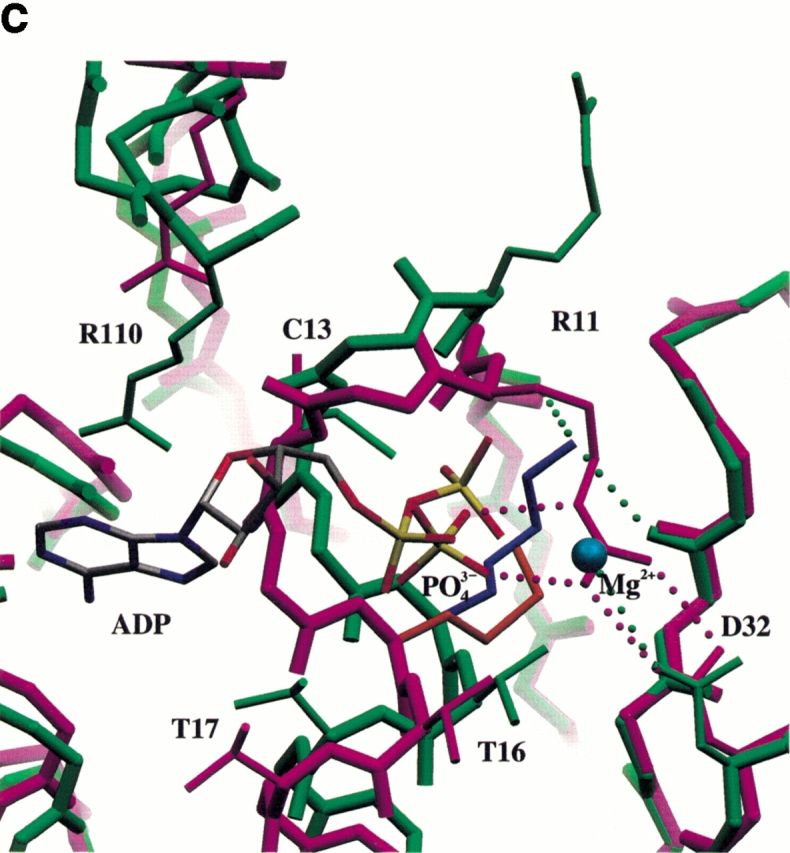
Superimposition of wild-type and K15M shikimate kinase after aligning the central sheet of 5 parallel β-strands using the program LSQKAB from the CCP4 suite. (a) A stereo view of the Cα superposition of the native SK structure (blue) and the K15M mutant (green). The ADP (colored according to atom type) and phosphate ion (cyan) of the two respective structures are shown. Key hinge regions in the shikimate-binding domain and lid domain of SK are highlighted with a magenta point and labeled. The key points of movement in the P-loop caused by the K15M mutation are highlighted with a red point and labeled. (b) Differences in Cα positions as a function of the residue number. Residues 113–122 are missing from the wild-type structure, and residues 1, 2, and 171–173 are missing from the mutant structure, owing to poor or ambiguous electron density. (c) View of the P-loop area of wild-type (in green) and K15M (in purple) shikimate kinase generated using SETOR (Evans 1993). The wild-type enzyme contains ADP and Mg2+ (blue sphere); the mutant enzyme contains a bound phosphate ion. K15 and M15 are shown in blue and orange, respectively. Significant hydrogen bonds are shown as dotted lines.
Both the increase in Tm and the higher ΔH for unfolding at higher temperatures (Fig. 2 ▶; Table 2) in the presence of substrate ligands—shikimate, ADP, and ATP—are consistent with stabilization of the native fold by specific ligand binding to the native state (Abele and Schulz 1995; Cooper 1999). Each ligand appears to exhibit its stabilizing effect independently and, for the nonproductive ADP/shikimate mixture, the effects are roughly additive. In cases of weak binding such as this, the effects of ligand on Tm and ΔHcal may be analyzed to give estimates of the affinity of ligand for the native protein (Cooper and McAuley-Hecht 1993). Application of this method to the DSC data observed here gives values for the dissociation constants for the binding of nucleotides to native SK in the range of 1.0–2.4 mM, which are consistent with values obtained by fluorescence quenching measurements (Table 1).
The X-ray structure of shikimate kinase mutant K15M
The structure of SK consists of a central sheet of 5 parallel β-strands, which are flanked by α-helices with the same overall topology as the nucleoside monophosphate (NMP) kinase family of proteins (Krell et al. 1998b). These proteins are characterized by large hinged movements of two regions of the structure around a core nucleotide-binding domain. By analogy with AK, the lid domain is thought to close over bound ATP. Similarly, the shikimate-binding domain, which is in the same overall position to the AMP-binding site of AK, is thought to undergo a significant movement associated with the binding of shikimate.
The structure of the K15M mutant has been solved by molecular replacement using wild-type SK as a model. Details of data collection, refinement, and final model statistics are shown in Table 3. The structure has been refined to 1.8 Å resolution, and a section of the final 2F0–Fc map is shown in Figure 3 ▶. The overall molecular architecture of wild-type and mutant SK is the same, and the central sheets of 5 β-strands (Fig. 4a ▶) can be closely superimposed (Fig. 5a ▶). A graph showing the r.m.s. deviation of Cα distance differences as a function of the residue number is shown in Figure 5b ▶. The superimposition of the 5 β-strands results in a r.m.s. difference of 0.20 Å for the Cα atoms and 0.55 Å for all protein atoms. This is essentially a perfect alignment because observed differences are the same order as the coordinate error of 0.18 Å, estimated by the method of Cruickshank (1999). However, in this alignment the overall difference in all Cα positions of 2.40 Å reflects very significant changes in conformation. These structural changes are most prominent in the P-loop, the shikimate-binding domain, and the region around the lid domain (Fig. 5b ▶).
Table 3.
Data collection, refinement, and final model statistics for the K15M mutant shikimate kinase
| Data | |
| X-ray source | SRS Daresbury, beamline 9.6 |
| Space group | P21 |
| Unit cell | a = 42.86 Å, b = 106.94 Å, c = 42.75 Å α = γ = 90.0°, β = 119.96° |
| Resolution | 25.0–1.8 Å |
| Total number of reflections | 164, 162 |
| Multiplicity | 5.3 |
| Completeness (%) (1.86–1.80 Å) | 93.0 (69.1) |
| Rmerge (%) (1.86–1.80 Å) | 7.8 (34.8) |
| Refinement | |
| Resolution | 25.0–1.8 Å |
| Rwork | 18.8% |
| Rfree | 22.7% |
| No. of unique reflections | 30,838 |
| Model | |
| No. of enzyme molecules | 2 |
| No. of amino acids | 340 |
| No. of heteroatoms | 68 (7 × MPD, 2 × PO4, 2 × Cl−) |
| No. of water molecules | 215 |
| r.m.s. deviation | |
| Bond length | 0.015 Å |
| Bond angle | 2.20° |
| Ramachandran quality, % in | |
| Most favored region | 93.2 |
| Allowed regions | 6.1 |
Fig. 3.
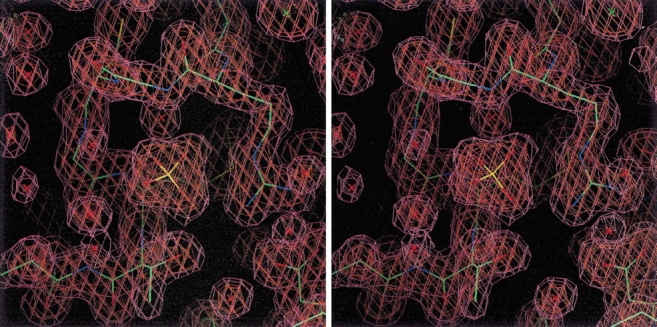
A stereo view of a representative section of the final weighted 2F0–Fc map of the K15M mutant of shikimate kinase contoured at 1 and 2 σ above the mean electron density. The region shown corresponds to amino acid residues 9–17 of the P-loop. The bound phosphate ion, water molecules (red crosses), and a chloride ion (green cross) are also shown.
Fig. 4.
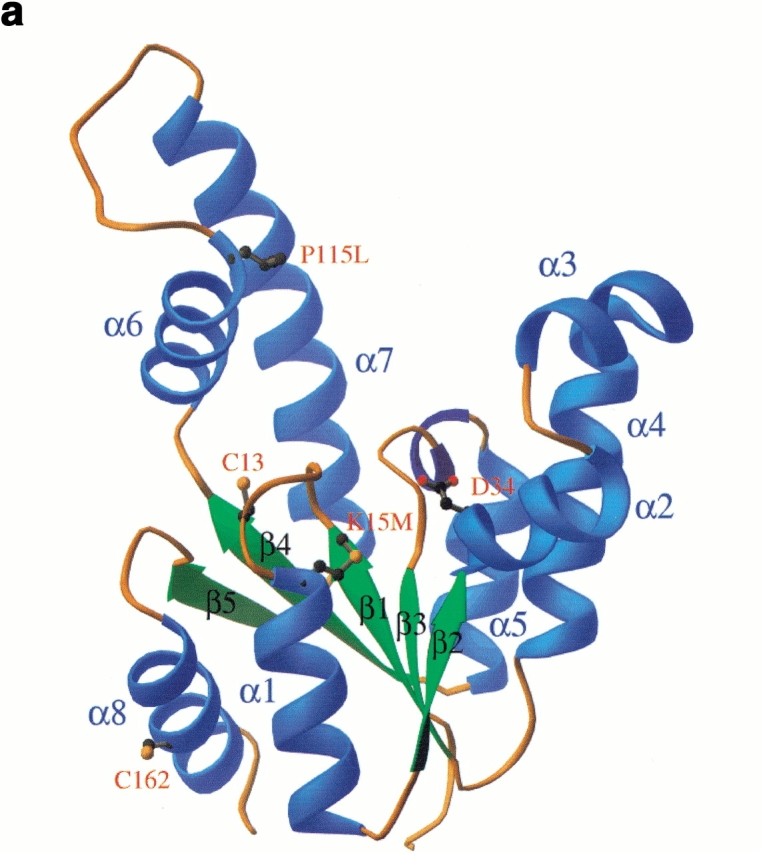

The three-dimensional structure of shikimate kinase mutant K15M. (a) A ribbon representation of the enzyme monomer colored according to secondary structure, with the position of residues mutated in this study shown in ball and stick. (b) Ribbon representation of the two independent molecules in the asymmetric unit. The extensive contacts of neighboring lid domains lead to a stabilization of this part of the molecule that is not visible in the native crystal structure. Both diagrams were generated using RIBBONS (Carson 1991).
In interpreting these changes it is important to take into account that the K15M mutant has crystallized in an open conformation equivalent to an apo-enzyme structure in which neither shikimate nor ADP would be bound.
SK and AKs have been shown to undergo structural changes upon substrate binding (Schulz et al. 1990; Krell et al. 1998b). These changes were most prominent in the lid domain that closes over bound nucleotide (Figs. 4, 5a ▶ ▶). This domain was disordered in wild-type SK, which as a consequence did not allow the inclusion of residues 113–122 in the model. K15M SK has crystallized in a different crystal form and contains the complete structure. Helix α6 is continued to A117 where a 6-residue lid domain with an open conformation joins to helix α7 at residue L123 (Fig. 4 ▶). Within the crystal, helices α6 and α7 and the lid domain of the two monomers form extensive interactions that stabilize the conformation of this region (Fig. 4b ▶). An unwanted point mutation in the lid domain (P115L) was detected during refinement of the model. (Although the DNA encoding the K15M mutant had been sequenced, the mutation had not been noted owing to the poor quality of the sequence data in this region.) This proline residue, which is conserved in the five known sequences of SK IIs but not in SK Is, is present in a region of the lid domain not seen in the X-ray structure of the wild-type enzyme. Its location at the end of helix α6 is shown in Figure 5a ▶; conceivably the P115L mutation has played a role in obtaining the new crystal form of SK. The mutation appears to have little effect on the overall conformation of the lid domain, which is very similar in structure to the short lid domains of a number of NMP kinases such as uridylate kinase and adenylate kinase isoenzyme 1 (Müller-Dieckmann and Schulz 1994).The orientation of the region around the lid domain (helices α6 and α7) differs from the native structure because of hinge movements at A102 and R139. These result in an ∼30° rotation of helices α6 and α7 (Fig. 5a ▶) up and away from the P-loop and also an ∼20° rotation in a perpendicular direction, moving the lid back from the P-loop in a horizontal direction. Interestingly, these hinges in position 102 and 139 are in exactly in the same structural position as the joints I and IV identified for AKs (Gerstein et al. 1993). The concerted movements in SK account for the large movements of residues such as L111, which is more than 8 Å away from its position in the native structure. It is likely that the observed structural changes in this region of the SK mutant enzyme are partially a result of absence of ligands in the structure, supported by favorable crystal contacts between the two molecules in the asymmetric unit, rather than a direct effect of the K15M mutation.
The second region, which is significantly different from the wild-type enzyme, is the shikimate-binding domain. This domain has been displaced by between 2 and 4 Å from the plane of the β-sheet by hinge movements at I35 and R59, resulting in a rotation of ∼15° (Fig. 5a ▶). In the mutant structure there is no electron density for shikimate in the active site, which suggests that this modified conformation is a more open structure and not correct for shikimate binding. The overall temperature factors for this domain are much lower in the mutant structure owing to the more extensive nature of the crystal contacts in this region when compared to those formed in the native structure (data not shown). Therefore, the differences in the shikimate-binding domain are attributable primarily to crystal contacts stabilizing a conformation that does not bind shikimate significantly and do not appear to be caused by the K15M mutation.
The P-loop itself is the third site that differs substantially from the wild-type enzyme (Fig. 5a ▶). In both SK structures, this region is not involved in crystal contacts, and the binding of different ligands in the P-loop of native SK has no effect on its molecular architecture (Krell et al. 1998b). Although K15M was co-crystallized with shikimate and ADP and these ligands were also present in the solution used for cryo-protection, none of these ligands were found in the molecular structure. Instead, a phosphate ion is bound to the P-loop (Fig. 5c ▶). As explained above, because the protein is in an open, apo-enzyme conformation, we would not expect to see either ligand (ADP or shikimate) present. We have shown that both these ligands bind to the mutant enzyme (Table 1) in a standard buffer system (see Materials and Methods). The phosphate ion in the mutant structure is positioned in a similar way as the β-phosphate of ADP in the wild-type protein, but has moved by ∼1 Å with respect to the core of strands (Fig. 5c ▶). If the β-phosphate of ATP were bound in the same position as the phosphate ion in the mutant structure, it is clear that the γ-phosphate would not be in the correct position to transfer to shikimate. The effect of the K15M mutation on the P-loop and helix α1 begins after G9; the loop is more bent down and shifted away from the plane of the β-sheet and down by ∼2 Å (Fig. 5a ▶). This shift is reduced to a 1-Å shift downward in the helix after G19 and continues until Y28. In the native structure K15 makes two good hydrogen bonds with the carbonyls of G9 and T77 of 2.6 Å and 2.8 Å respectively. These hydrogen bonds are lost, and as a result the carbonyl oxygen of G9 shifts down 0.5 Å relative to the native structure. Contrary to expectations from modeling using Quanta (Molecular Simulations), methionine cannot be accommodated in a similar manner to lysine at position 15 without perturbing the main-chain conformation of helix α1. The shift of 1.2 Å in its Cα position between mutant and wild-type enzyme (Fig. 5b ▶), away from the plane of the β-sheet, clearly reflects this.
In detail, R11 is one of the first residues in the P-loop to be significantly affected by the mutation (Fig. 5c ▶). It is poorly ordered in wild-type structures and makes a weak interaction with E131. In the K15M mutant R11 forms a strong ion pair with D32, the residue shown to be one of the ligands of the nucleotide-bound magnesium ion (Krell et al. 1998b). The formation of this ion pair is most likely the reason for the increased thermal stability of this mutant as expressed by the increase in melting temperature by 4.2°C (Table 2). The guanidinium group of R11 in the mutant protein is not only in a very similar position to the bound magnesium ion in the wild-type protein but also forms two hydrogen bonds with the phosphate bound in the P-loop of 2.9 Å and 3.1 Å length. Conformational changes within the P-loop result in the side chain of C13 being unable to adopt its conformation in the center of the P-loop "anion hole" owing to steric reasons (Fig. 5c ▶). Instead, the side chain adopts a more favorable rotamer (as seen in 60% of cysteine residues as opposed to 14% in the native SK conformation; data from Quanta), and is moved out of the P-loop, forming an interaction with the side chain of R110. The results from the chemical modification experiments indicate the presence of an ion pair in the K15M mutant, which is consistent with the proposal that the perturbation of the P-loop occurs in solution as well as in the crystal. Other key residues in the Walker A motif, namely, T16 and T17, which are important in the coordination of the ATP ribose, are shifted nearly 2 Å away from D32, which is involved in the coordination of Mg2+.
These data suggest that K15 in SK plays an important structural role in maintaining the conformation of the P-loop. This is consistent with site-directed mutagenesis studies of the P-loop lysine in other proteins, including AK from muscle (Tian et al. 1990; Byeon et al. 1995) and E. coli (Reinstein et al. 1990), adenylosuccinate synthetase (Kang et al. 1997) and the vacuolar ATPases (Liu et al. 1997), all of which have shown that replacement of the lysine leads to structural perturbations. In contrast, the replacement of other P-loop residues that occur on the N-terminal side of the lysine in AK (Müller and Schulz 1993) and oncogenic p21ras (Taiz et al. 1994; Liu et al. 1997; Hunke and Schneider 1999) do not seem to have such strong structural consequences.
We conclude that K15 is a critical residue in shikimate kinase, not only as a residue that stabilizes the pentavalent transition state of the γ-phosphoryl group during the phosphotransfer reaction, but also because it plays a key role in maintaining the conformation of the P-loop.
Conclusions
Several conclusions emerge from this study of shikimate kinase mutated in the P-loop region of the structure. The side chain of K15 has both an essential structural and a mechanistic role, whereas the thiol of C13 is not essential for the activity of the enzyme. The other mutants confirm that D34 is important for enzyme stability and in addition may have a functional role, whereas C162 is of no importance. Direct measurements of substrate binding together with the steady-state kinetic studies provide evidence of synergy in substrate binding. This is consistent with the earlier CD studies, which demonstrate significant changes in secondary structure accompanying the binding of either substrate (Krell et al. 1998b). The P-loop lysine (K15) shows very markedly enhanced chemical reactivity toward TNBS, which appears to be owing to the proximity of the side chain of C13. The reactivity of this cysteine has been probed using 2PDS, giving evidence for an ion pair between C13 and K15 in the wild-type enzyme. Studies of the wild-type enzyme and the mutants C13S and K15M suggest that the enhanced reactivity of K15 to TNBS in the wild type may be caused by rapid modification of C13 followed by intramolecular transfer of the TNBS group to the lysine residue. It is unlikely that the enhanced reactivity of K15 can be associated with an essential catalytic role for this residue, in view of the fact that the C13S mutant (which does not show the enhanced reactivity) retains most of the activity of wild-type enzyme. Finally, the structural changes accompanying the K15M mutation leads to a significant enhancement of enzyme stability caused by the formation of an unpredicted arginine–aspartate salt bridge.
Materials and methods
Protein expression, purification, and estimation of protein concentration
Wild-type and mutant SK from Erwinia chrysanthemi were overexpressed in Escherichia coli BL21(DE3)pLysS using a T7 RNA polymerase expression system (Studier and Moffatt 1986). Proteins were purified using anion exchange chromatography on DEAE Sepharose, hydrophobic interaction chromatography on phenyl Sepharose, and gel filtration as described in Krell et al. (1997). Throughout purification, protein was detected using the enzyme assay, apart from inactive mutants K15M and D34N, which were detected using SDS-PAGE analysis. Protein concentrations were determined using the method of Bradford (1976) or Lowry et al. (1951) with bovine serum albumin (BSA) as a standard.
The enzyme assay
SK was assayed at 25°C by coupling the release of ADP to the oxidation of NADH using pyruvate kinase (EC 2.7.1.40) and lactate dehydrogenase (EC 1.1.1.27) as coupling enzymes. Shikimate-dependent oxidation of NADH was monitored at 340 nm (ɛ = 6180 M−1 cm−1). The assay mixture contained 50 mM triethanolamine hydrochloride/KOH buffer at pH 7.0, 50 mM KCl, 5 mM MgCl2, 1.6 mM shikimic acid, 5 mM ATP, 1 mM phosphoenolpyruvate, 0.1 mM NADH, 3 units of pyruvate kinase/mL, and 2.5 units of lactate dehydrogenase/mL. Kinetic parameters were estimated using nonlinear regression to the Michaelis–Menten equation (Microcal Software); the errors in the parameters were less than 5%.
Site-directed mutagenesis
DNA fragments coding for the mutant enzyme were prepared following the two-step PCR method of Higuchi et al. (1988) using Vent DNA polymerase (New England Biolabs) and appropriate buffers. The following mismatch oligonucleotides were used for the PCR amplification: K15M, GGGTGCGGAATGACCACCGT CGGC; C13S, GGCGCCAGAGGGAGCGGAAAAACC; C162S, CCCGCCGCGATTGTCAGCGAATTGATG; and D34N, GGCT ATGAGTTTGTCGATACGAATATTTTTATGCAG. DNA fragments obtained from the PCR were purified using the PCR Clean Up Kit (Promega) and inserted into the dephosphorylated vector pTB361 (Zeneca, Patent application no. 92301456.8) using the NdeI and BglII restriction sites. Competent E. coli BL21(DE3)pLysS were transformed with the ligation mixes, and colonies were selected on chloramphenicol- and tetracyclin-containing LB-agar plates.
Chemical modification with 2,4,6-trinitrobenzenesulfonic acid (TNBS)
Enzyme modification was carried out in a volume of 2 mL at 25°C with constant stirring. The inactivation was carried out in the dark. The enzyme (4 μg/mL) was preincubated for 5 min in 50 mM sodium borate buffer at pH 9.2. TNBS stock solutions (500 μM for experiments with the native enzyme, 50 mM for the C13S mutant) were prepared in borate buffer, and the pH was readjusted to 9.2 using concentrated NaOH. Aliquots were added to a final concentration of 0.5–2.5 μM for the native enzyme and 0.25–2.0 mM for the mutant enzyme. For substrate protection experiments, 50 mM stock solutions of shikimate and ATP were prepared in borate buffer and the pH was readjusted to 9.2. During the inactivation reaction, aliquots were taken at the stated times for assay of remaining enzyme activity. The percentage of inactivation was calculated from the ratio of enzyme activity after a certain time of treatment to the initial enzyme activity.
Reactivity probe kinetics using 2,2`-dipyridyl disulfide (2PDS)
All reactions were carried out at 25°C and I 0.1 M in aqueous buffers containing 1 mM EDTA and 2.5% (v/v) glycerol under pseudo first-order conditions with [2PDS] |L: [enzyme], using a Hi-Tech Scientific SF-61 stopped-flow (SF) spectrophotometer, kinetics workstation, and data acquisition and analysis software. 2PDS was the Aldrich product, purified by recrystallization (Pinitglang et al. 1997). The enzymes (SK and its K15M and C162S mutants) were incubated with l-cysteine (20 mM) in Tris/HCl buffer at pH 8.0, I 0.1 M, containing EDTA (0.5 mM) for 30 min to reduce any reversibly oxidized protein. The enzymes were then freed from low Mr material by gel filtration through a column of Sephadex G-25 (20 × 2.5 cm) using 0.1 M KCl containing 3 mM KH2PO4 at pH 7.35 as eluent. Concentrations in the reaction chamber were [E] = 3.0–5.5 μM and [2PDS] = 0.23–0.3 mM.
Reactions were monitored at 343 nm, and release of the chromophoric product, pyridine-2-thione, was quantified by using Δɛ343 = 8.08 × 103 M−1 cm−1, which is pH-independent in acidic media (Stuchbury et al. 1975). Observed first-order rate constants (kobs) were obtained by fitting the absorbance (Abs) versus t data collected by the microcomputer of the SF-system to the equation for a single exponential process. Values of second-order rate constants (k) were calculated from k = kobs/[2PDS]. pH-dependent kinetic data were evaluated and displayed as described by Pinitglang et al. (1997), making use initially of SKETCHER (Brocklehurst and Topham 1990; Topham et al. 1991) and subsequently of SIGMAPLOT 5.0. SKETCHER permits rapid estimation of characterizing parameters (pKa values and values of pH-independent rate constants, k̃) by interactive manipulation of calculated curves.
Circular dichroism (CD) and fluorescence measurements
Protein samples were dialyzed into 35 mM Tris/HCl at pH 7.4, 5 mM KCl, 2.5 mM MgCl2, and 0.4 mM DTT (buffer A). CD measurements were made at 20°C using a JASCO J-600 spectropolarimeter. Protein concentrations and cell pathlengths were 1 mg/mL and 0.5 cm for near-UV and 0.2–0.5 mg/mL and 0.02 cm for far-UV CD studies.
Fluorescence measurements were made at 20°C using a Perkin-Elmer LS50B spectrofluorimeter. The enzyme concentration for each experiment was 0.2 mg/mL in a cuvette of 1 mL volume. The excitation wavelength was 295 nm, and emissions were recorded at 350 nm. In experiments where the binding of nucleotide was studied, correction of the inner filter effect was made using N-acetyltryptophan amide as a model compound as described by Price (1972).
Differential scanning calorimetry (DSC)
Experiments to study thermal unfolding of shikimate kinase in solution were carried out using a Microcal MCS-DSC system (Microcal) under standard operating conditions (Cooper and Johnson 1994). Protein samples were exhaustively dialyzed into buffer A, and the protein concentration was adjusted to 1 mg/mL by dilution with dialysis buffer. All samples were degassed gently at room temperature prior to loading into the DSC cell. Stock solutions of ligands were prepared using the same dialysis buffer, and the pH was carefully adjusted back to pH 7.5 when necessary, using concentrated KOH. DSC data were collected over a temperature range of 20–90°C at a nominal scan rate of 60°C/h. Data were analyzed using the standard Microcal Origin software package (Microcal Software).
Crystallization and structure solution of shikimate kinase K15M
The enzyme was exhaustively dialyzed into 10 mM Tris/HCl at pH 7.5, 0.5 mM DTT, and subsequently concentrated using Centricon-10 centrifugal concentrators (Amicon, Stonehouse) to a concentration of 15 mg/mL. Shikimate and ADP (freshly made up solution in water, pH adjusted to 7.5) were added to a final concentration of 2.5 mM, and MgCl2 was added to a final concentration of 10 mM. The pH was readjusted to pH 7.5 and the protein used for crystallization was ∼8–10 mg/mL. Crystals were obtained using sitting-drop vapor diffusion, mixing 2 μL of protein with an equal volume of reservoir solution (10% PEG 8000, 100 mM Tris/HCl at pH 8.0).
Details of data collection, refinement, and final model statistics are shown in Table 3. Initial attempts to cryo-cool crystals using the conditions used for the native enzyme (17.5 % glycerol in artificial mother liquor; Krell et al. 1997) failed, and crystals seemed to shatter under these conditions. Instead, a cryo-protectant containing 15% 2-methyl-2,4-pentanediol (MPD) in artificial mother liquor with 2.5 mM shikimate, 2.5 mM ADP, and 10 mM MgCl2 was successful; crystals diffracted at 100 K to 1.8 Å on station 9.6 at the Daresbury synchrotron radiation source (SRS).
Data were processed using DENZO and scaled with SCALEPACK (Otwiniwski 1993). The data were initially processed and scaled in orthorhombic spacegroup C2221, unit cell dimensions a = 42.75, b = 74.27, c = 106.92 Å, α = β = γ = 90°, which has a packing density of 2.2 Å3 D−1, corresponding to one molecule in the asymmetric unit. There were a significant number of rejections in this spacegroup, coupled with a modest deterioration in the merging R-factor statistics; therefore, the structure was finally processed in monoclinic P21 with presumably two molecules in the asymmetric unit (Table 3). The structure was solved by molecular replacement with the structure of the native enzyme (pdb entry 2shk) using AMoRe (Navaza 1994). Searching for two molecules in the asymmetric unit, there was a clear solution with a correlation coefficient of 76.4% and an R factor of 31.1%. The model was built using the program Quanta (Molecular Simulations) and refined using REFMAC (Murshudov et al. 1996) in combination with ARP (Lamzin and Wilson 1993). Tight NCS restraints were maintained throughout refinement. A section of the final 2F0 − Fc electron density map can be seen in Figure 3 ▶. The refined coordinates and structure factors have been deposited at the Protein Data Bank with the accession codes 1e6c and r1e6csf.
Acknowledgments
We are grateful to Margaret Nutley for technical assistance and the BBSRC for funding. The Biological Microcalorimetry Facility in Glasgow is jointly funded by BBSRC and EPSRC.
The publication costs of this article were defrayed in part by payment of page charges.This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AK, adenylate kinase
CD, circular dichroism
DSC, differential scanning calorimetry
DTT, dithiothreitol
k, second-order rate constant
kobs, (pseudo) first-order rate constant
MPD, 2-methyl-2,4-pentanediol
NCS non-crystallographic symmetry
2PDS, 2,2`-dipyridyl disulfide
P-loop, phosphate binding loop
r.m.s., root mean square
SF, stopped-flow
SK, shikimate kinase
TNBS, 2,4,6-trinitrobenzenesulfonic acid
UV, ultraviolet
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/
References
- Abele, U. and Schulz, G.E. 1995. High-resolution structure of adenylate kinase from yeast ligated with inhibitor Ap5A, showing the pathway of phosphoryl transfer. Protein Sci. 4 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkett, D.J., Price, N.C., Radda, G.K., and Salmon, A.G. 1970. The reactivity of SH groups with a fluorogenic reagent. FEBS Lett. 6 346–348. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 255–260. [DOI] [PubMed] [Google Scholar]
- Brocklehurst, K. 1982. Two-protonic-state electrophiles as probes of enzyme mechanism. Methods Enzymol. 87C 427–469. [DOI] [PubMed] [Google Scholar]
- ———. 1994. A sound basis for pH-dependent kinetic studies on enzymes. Protein Eng. 7 291–299. [DOI] [PubMed] [Google Scholar]
- ———. 1996. pH-dependent kinetics. In Enzymology Labfax (ed. P.C. Engel), pp. 175–198. Academic Press, San Diego, CA.
- Brocklehurst, K., and Topham, C.M. 1990. Kinetic-parameters of the acyl-enzyme mechanism and conditions for quasi-equilibrium and for optimal catalytic characteristics. Biochem. J. 270 561–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocklehurst, K., Watts, A.B., Patel, M., Verma, C., and Thomas, E.W. 1998. Cysteine proteinases. In Comprehensive Biological Catalysis (ed. M.L. Sinnott), pp. 381–423. Academic Press, London.
- Brocklehurst, S.M., Topham, C.M., and Brocklehurst, K. 1990. A general kinetic equation for multihydronic state reactions and rapid procedures for parameter evaluation. Biochem. Soc. Trans. 18 598–599. [DOI] [PubMed] [Google Scholar]
- Byeon, I.-J.L., Shi, Z., and Tsai, M.-D. 1995. Mechanism of adenylate kinase—The essential lysine helps to orient the phosphates and the active-site residues to proper conformations. Biochemistry 34 3172–3182. [DOI] [PubMed] [Google Scholar]
- Carson, M. 1991. Ribbons 2.0. J. Appl. Cryst. 24 958–961. [Google Scholar]
- Coggins, J.R. 1989. The shikimate pathway as a target for herbicides. In Herbicides and plant metabolism (ed. A. Dodge), pp. 96–112. Cambridge University Press, Cambridge, UK.
- Cooper, A. 1999. Thermodynamics of protein folding and stability. In Protein: A comprehensive treatise (ed. G. Allen), Vol. 2, pp. 217–270. JAI Press, Stamford, CT.
- Cooper, A. and Johnson, C.M. 1994. Isothermal titration microcalorimetry. In Methods in molecular biology, Vol. 22: Microscopy, optical spectroscopy, and macroscopic techniques, (eds. C. Jones et al.), pp. 125–136. Humana Press, Totowa, NJ. [DOI] [PubMed]
- Cooper, A. and McAuley-Hecht, K.E. 1993. Microcalorimetry and the molecular recognition of peptides and proteins. Phil. Trans. R. Soc. Lond. A345 25–35. [Google Scholar]
- Cruikshank, D.W.J. 1999. Remarks about protein structure precision. Acta Crystal. D 55 583–601. [DOI] [PubMed] [Google Scholar]
- Davies, G.M., Barrett-Bee, K.J., Jude, D.A., Lehan, M., Nichols, W.W., Pinder, P.E., Thain, J.L., Watkins, W.J., and Wilson, R.G. 1994. (6S)-6-Fluoroshikimic acid, an antimicrobial agent acting on the shikimate pathway. Antimicrob. Agents and Chemother. 38 403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feyter, R.C. and Pittard, J. 1986. Purification and properties of shikimate kinase II from Escherichia coli K-12. J. Bacteriol. 165 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Feyter, R.C., Davidson, B.E., and Pittard, J. 1986. Nucleotide sequences of the transcription unit containing the aroL and aroM genes from Escherichia coli K-12. J. Bacteriol. 165 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delepelaire, P. 1994. PRTD, the integral membrane ATP-binding cassette component of the Erwinia chrysanthemi metalloprotease secretion system, exhibits a secretion signal-regulated ATPase activity. J. Biol. Chem. 269 27952–27957. [PubMed] [Google Scholar]
- Deyrup, A.T., Krishnan, S., Cockburn, B.N., and Schwartz, N.B. 1998. Deletion and site-directed mutagenesis of the ATP-binding motif (P-loop) in the bifunctional murine ATP-sulfurylase/adenosine 5`-phosphosulfate kinase enzyme. J. Biol. Chem. 273 9450–9456. [DOI] [PubMed] [Google Scholar]
- Dixon, H.B.F. 1973. Shapes of curves of pH-dependence of reactions. Biochem. J. 131 149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely, B. and Pittard, J. 1979. Aromatic amino acid biosynthesis: Regulation of shikimate kinase in Escherichia coli K-12. J. Bacteriol. 138 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, S.V. 1993. SETOR: Hardware lighted three-dimensional solid model representations of macromolecules. J. Mol. Graphics 11 134–138. [DOI] [PubMed] [Google Scholar]
- Font, B. and Gautheron, D.C. 1980. General and kinetic properties of pig heart mitochondrial adenylate kinase. Biochim. Biophys. Acta 611 299–308. [DOI] [PubMed] [Google Scholar]
- Freedman, R.B. and Radda, G.K. 1968. The reaction of 2,4,6-trinitrobenzenesulphonic acid with amino acids, peptides and proteins. Biochem. J. 108 363–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein, M., Schulz, G.E., and Chotia, C. 1993. Domain closure in adenylate kinase: Joints on either side of two helices close like neighbouring fingers. J. Mol. Biol. 229 494–501. [DOI] [PubMed] [Google Scholar]
- Goldfarb, A.R. 1966. A kinetic study of the reactions of amino acids and peptides with trinitrobenzenesulfonic acid. Biochemistry 5 2570–2574. [DOI] [PubMed] [Google Scholar]
- Goldin, B.R. and Frieden, C. 1971. Effects of trinitrophenylation of specific lysyl residues on the catalytic, regulatory and molecular properties of bovine liver glutamate dehydrogenase. Biochemistry 10 3527–3534. [DOI] [PubMed] [Google Scholar]
- Higuchi, R., Krummel, B., and Saiki, R.K. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments—Study of protein and DNA interactions. Nucl. Acids Res. 16 7351–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishida, T., Iwasaki, H., Yagi, T., and Shinagawa, H. 1999. Role of Walker motif A of RuvB protein in promoting branch migration of Holliday junctions—Walker motif A mutations affect ATP binding, ATP hydrolyzing, and DNA binding activities of RuvB. J. Biol. Chem. 274 25335–25342. [DOI] [PubMed] [Google Scholar]
- Hunke, S. and Schneider, E. 1999. A Cys-less variant of the bacterial ATP binding cassette protein MalK is functional in maltose transport and regulation. FEBS Lett. 448 131–134. [DOI] [PubMed] [Google Scholar]
- Hyde, S.C., Emsley, P., Hartshorn, M.J., Mimmack, M.M., Gileadi, U., Pearce, S.R., Gallagher, M.P., Gill, D.R., Hubbard, R.E., and Higgins, C.G. 1990. Structural model of ATP-binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 346 362–365. [DOI] [PubMed] [Google Scholar]
- Kang, C.H., Sun, N., Poland, B.W., Gorrell, A., Honzatko, R.B., and Fromm, H.J. 1997. Residues essential for catalysis and stability of the active site of Escherichia coli adenylosuccinate synthetase as revealed by directed mutation and kinetics. J. Biol. Chem. 272 11881–11885. [DOI] [PubMed] [Google Scholar]
- Krell, T., Coyle, J.E., Horsburgh. M.J., Coggins, J.R., and Lapthorn, A.J. 1997. Crystallization and preliminary X-ray crystallographic analysis of shikimate kinase from Erwinia chrysanthemi. Acta Crystal. D 53 612–614. [DOI] [PubMed] [Google Scholar]
- Krell, T., Chackrewarthy, S., Pitt, A.R., Elwell, A., and Coggins, J.R. 1998a. Chemical modification monitored by electrospray mass spectrometry: A rapid and simple method for identifying and studying functional residues in enzymes. J. Peptide Res. 51 201–209. [DOI] [PubMed] [Google Scholar]
- Krell, T., Coggins, J.R., and Lapthorn, A.J. 1998b. The three-dimensional structure of shikimate kinase. J. Mol. Biol. 278 983–997. [DOI] [PubMed] [Google Scholar]
- Lamzin, V.S. and Wilson, K.S. 1993. Automated refinement of proteins. Acta Crystal. D 49 129–147. [DOI] [PubMed] [Google Scholar]
- Liu, Q., Leng, X.H,, Newman, P.R., Vasilyeva, E., Kane, P.M., and Forgac, M. 1997. Site-directed mutagenesis of the yeast V-ATPase A subunit. J. Biol. Chem. 272 11750–11756. [DOI] [PubMed] [Google Scholar]
- Lowry, O., Rosebrough, N.J., Farr, A.L., and Randall, R.J. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193 265–275. [PubMed] [Google Scholar]
- Means, G.E. and Feeney, R.E. 1971. Chemical modification of proteins, see p. 217. Holden-Day, San Francisco, CA.
- Millar, G., Lewendon, A., Hunter, M.G., and Coggins, J.R. 1986. The cloning and expression of the aroL gene from Escherichia coli K-12. Biochem. J. 237 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, C.W. and Schulz, G.E. 1988. Structure of the complex of adenylate kinase from Escherichia coli with the inhibitor P1,P5-di(adenosine-5`-)pentaphosphate. J. Mol. Biol. 202 909–912. [DOI] [PubMed] [Google Scholar]
- ———. 1993. Crystal-structures of 2 mutants of adenylate kinase from Escherichia coli that modify the Gly-loop. Proteins Struct. Funct. Genet. 15 42–49. [DOI] [PubMed] [Google Scholar]
- Müller-Dieckmann, H.-J. and Schulz, G.E. 1994. The structure of uridylate kinase with its substrates, showing the transition state geometry. J. Mol. Biol. 236 361–367. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Dodson, E.J., and Vagin, A.A. 1996. Application of maximum likelihood methods for macromolecular refinement. In Macromolecular refinement (eds. E. Dodson et al.), pp. 93–104. SERC Daresbury Laboratory, Warrington, UK.
- Otwiniwski, Z. 1993. Oscillation data reduction program. In Data collection and processing (eds. L. Sawyer et al.), pp. 56–62. SERC Daresbury Laboratory, Warrington, UK.
- Pai, E.F., Kabsch, W., Krengel, U., Holmes, K.C., John, J., and Wittinghofer, A. 1989. Structure of the guanine-nucleotide-binding domain of the HA-ras oncogene product p21 in the triphosphate conformation. Nature 341 209–214. [DOI] [PubMed] [Google Scholar]
- Pinitglang, S., Watts, A.B., Patel, M., Reid, J.D., Noble, M.A., Gul, S., Bokth, A., Naeem, A., Patel, H., Thomas, E.W., et al. 1997. A classical enzyme active centre motif lacks catalytic competence until modulated electrostatically. Biochemistry 36 9968–9982. [DOI] [PubMed] [Google Scholar]
- Plou, F.J., Kowlessur, D., Malthouse, J.P.G., Mellor, G.W., Hartshorn, M.J., Pinitglang, S., Patel, H., Topham, C.M., Thomas, E.W., Verma, C., et al. 1996. Characterization of the electrostatic perturbation of a catalystic site (Cys)-S−/(His)-Im+H ion-pair in one type of serine proteinase architecture by kinetic and computational studies on chemically-mutated subtilisin variants. J. Mol. Biol. 257 1088–1111. [DOI] [PubMed] [Google Scholar]
- Ponder, J.W. and Richards, F.M. 1987. Tertiary templates for proteins—Use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 193 775–793. [DOI] [PubMed] [Google Scholar]
- Price, N.C. 1972. The interaction of nucleotides with kinases, monitored by changes in protein fluorescence. FEBS Lett. 24 21–23. [DOI] [PubMed] [Google Scholar]
- Reinstein, J., Schlichting, I., and Wittinghofer, A. 1990. Structurally and catalytically important residues in the phosphate binding loop of adenylate kinase of Escherichia coli. Biochemistry 29 7451–7459. [DOI] [PubMed] [Google Scholar]
- Rhoads, D.G. and Lowenstein, J.M. 1968. Initial velocity and equilibrium kinetics of myokinase. J. Biol. Chem. 243 3963–3972. [PubMed] [Google Scholar]
- Schulz, G.E., Müller, C.W., and Diederichs, K. 1990. Induced-fit movements in adenylate kinases. J. Mol. Biol. 213 627–630. [DOI] [PubMed] [Google Scholar]
- Sigal, I.S., Gibbs, J.B., D'Alonzo, J.S., Temeles, G.L., Wolanski, B.S., Socher, S.H., and Scolnick, E.M. 1986. Mutant ras-encoded proteins with altered nucleotide binding exert dominant biological effects. Proc. Natl. Acad. Sci. USA 83 952–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C.A. and Rayment, I. 1996. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J. 70 1590–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spuergin, P., Abele, U., and Schulz, G.E. 1995. Stability, activity and structure of adenylate kinase mutants. Eur. J. Biochem. 231 405–413. [DOI] [PubMed] [Google Scholar]
- Stuchbury, T., Shipton, M., Norris, R., Malthouse, J.P.G., Brocklehurst, K., Herbert, J.A.L. and Suschitzky, H. 1975. A reporter group delivery system with both absolute and selective specificity for thiol groups and an improved fluorescent probe containing the 7-nitrobenzo-2-oxa-1,3 diazole moiety. Biochem. J. 151 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier, F.W. and Moffat, B.A. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189 113–130. [DOI] [PubMed] [Google Scholar]
- Suzuki, K., Mizuguchi, M., Gomi,T., and Itagaki, E.J. 1995. Identification of a lysine residue in the NADH-binding site of salicylate hydroxylase from Pseudomonas putida S-1. J. Biochem. (Tokyo) 117 579–585. [DOI] [PubMed] [Google Scholar]
- Taiz, L., Nelson, H., Maggert, K., Morgan, L., Yatabe, B., Taiz, S.L., Rubinstein, B., and Nelson, N. 1994. Functional analysis of conserved cysteine residues in the catalytic subunit of the yeast vacuolar H+-ATPase. Biochem. Biophys. Acta 1194 329–334. [DOI] [PubMed] [Google Scholar]
- Tian, G., Yan, H., Jiang, R.-T., Kishi, F., Nakazawa, A., and Tsai, M.-D. 1990. Mechanism of adenylate kinase. 6. Are the essential lysines essential? Biochemistry 29 4296–4304. [DOI] [PubMed] [Google Scholar]
- Topham, C.M., Salih, E., Frazao, C., Kowlessur, D., Overington, J.P., Thomas, M., Brocklehurst, S.M., Patel, M., Thomas, E.W., and Brocklehurst, K. 1991. Structure–function relationships in the cysteine proteinases actinidin, papain and papaya proteinase Ω deduced by knowledge-based modelling and active centre characteristics determined by two-hydronic-state reactivity probe kinetics and kinetics of catalysis. Biochem. J. 280 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinella, D., Gagny, B., Joseleau-Petit, D., D'Ardi, R., and Cashel, M. 1996. Mecillinam resistance in Escherichia coli is conferred by loss of a second activity of the aroK protein. J. Bacteriol. 178 3818–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, J.E., Saraste, M., Runswick, M.J., and Gay, N.J. 1982. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipp, M.J. and Pittard, A.J. 1995. A reassessment of the relationship between aroK- and aroL-encoded shikimate kinase enzymes of Escherichia coli. J. Bacteriol. 177 1627–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]