

Abstract
Shorthorn sculpins, Myoxocephalus scorpius, are protected from freezing in icy seawater by alanine-rich, α-helical antifreeze proteins (AFPs). The major serum isoform (SS-8) has been reisolated and analyzed to establish its correct sequence. Over most of its length, this 42 amino acid protein is predicted to be an amphipathic α-helix with one face entirely composed of Ala residues. The other side of the helix, which is more heterogeneous and hydrophilic, contains several Lys. Computer simulations had suggested previously that these Lys residues were involved in binding of the peptide to the {11–20} plane of ice in the <−1102> direction. To test this hypothesis, a series of SS-8 variants were generated with single Ala to Lys substitutions at various points around the helix. All of the peptides retained significant α-helicity and remained as monomers in solution. Substitutions on the hydrophilic helix face at position 16, 19, or 22 had no obvious effect, but those on the adjacent Ala-rich surface at positions 17, 21, and 25 abolished antifreeze activity. These results, with support from our own modeling and docking studies, show that the helix interacts with the ice surface via the conserved alanine face, and lend support to the emerging idea that the interaction of fish AFPs with ice involves appreciable hydrophobic interactions. Furthermore, our modeling suggests a new N terminus cap structure, which helps to stabilize the helix, whereas the role of the lysines on the hydrophilic face may be to enhance solubility of the protein.
Keywords: α-Helix, antifreeze, helix cap structure, molecular modeling, thermal hysteresis, van der Waals interactions
Antifreeze proteins (AFPs) adsorb to the surface of ice and inhibit the addition of water to the crystal (Raymond and DeVries 1977). At high AFP concentrations, this effect is sufficient to lower the nonequilibrium freezing point of fish blood below the freezing point of seawater and ensure the survival of the host in ice-laden marine environments. The forces involved in binding AFPs to ice are not well defined. The traditional view that a hydrogen bond match between AFP and ice dominates binding (DeVries 1983; Chou 1992; Wen and Laursen 1992) has been challenged recently (Chao et al. 1997; Haymet et al. 1998; Zhang and Laursen 1998). To understand AFP binding, it is necessary to delineate both the ice surfaces bound by AFP and the surface of the protein that binds to ice. The former is particularly well defined for type I AFP from winter flounder. This alanine-rich amphipathic helix binds to the {11–20} pyramidal planes on which it is aligned in the <−1102> direction (Knight et al. 1991; Laursen et al. 1994). The winter-flounder type I AFP has Thr/Asx residues projecting on one side of the helix with a regular 16.5Å spacing that matches the underlying ice lattice geometry (Sicheri and Yang 1995). It was suggested originally that the hydrophilic side binds to ice by hydrogen bonding (DeVries and Lin 1977), but amino acid replacements have cast doubt on the primacy of the Thr–OH group (Chao et al. 1997) as well as the role of Asx in ice binding (Loewen et al. 1999). The fact that valine is a better replacement for threonine than serine seems to emphasize the importance of the Thr–CH3 group. In the latter study, replacement of the Asx residues had little effect on antifreeze activity with the exception of Gln substitutions. The longer side chain of Gln could potentially interact with the neighboring Thr to alter its rotamer conformation.
Therefore, the ice-binding site of the α-helical antifreeze protein from winter flounder was re-evaluated by replacing individual alanines around the helix with leucine to act as steric mutations that would interfere with ice binding (Baardsnes et al. 1999). Leucines were well tolerated on the hydrophilic side, but completely eliminated ice binding when they protruded from the conserved alanine-rich surface adjacent to the line of Thr. This suggested that the ice-binding site of the flounder antifreeze is a relatively hydrophobic surface rotated ∼110° away from the traditional ice-binding site. In support of this result, Monte Carlo simulations of winter-flounder antifreeze docking to ice have indicated that this face can interact to the ice surface through van der Waals contributions (Dalal and Sönnichsen 2000).
Type I AFPs, classified by their alanine richness and α-helicity, are also found in some sculpins (Hew et al. 1980). It is not clear if they are homologs of the flounder type I AFPs. Although a tenuous claim to homology can be made through the flounder skin isoforms (Fletcher et al. 2001), fishes more closely related to sculpins than flounders are known to have different AFPs in their serum (Ewart et al. 1992; Deng et al. 1997). The prominent type I AFP serum isoform (SS-8) in the shorthorn sculpin (Myoxocephalus scorpius) has some notable differences from the flounder AFPs. It binds to the secondary prism planes of ice, {11–20} (Cheng and DeVries 1991). Also, it does not have Thr and Asx repeating at 11-residue intervals. It is, however, amphipathic, and its Lys-rich hydrophilic side has been modeled as the ice-binding site with the lysine residues providing specific contacts to ice (Wierzbicki et al. 1996). In view of the results with flounder AFP, we have set out to test whether the hydrophilic side is the ice-binding face of the helix.
Prior to attempting these structure-function studies, it was necessary to resolve discrepancies in the literature regarding the sequence of SS-8, the large AFP isoform in shorthorn sculpin serum. The original widely quoted sequence MN GETPAQKAARLAAAAALAAKTAADAAAKAAAKAA AIAAAAASA (Hew et al. 1985; Yang et al. 1988; Harding et al. 1999) is different from that listed in Cheng and DeVries (1991) and modeled by Wierzbicki et al. (1996), MDGETPAQKAARLAAAAAALAAKTAADAAAKAAA IAAAAASA. Another version of the latter sequence is missing the last two residues (Knight et al. 1991). There are several isoforms of type I AFP in both flounder and sculpin. To see whether the discrepancy was due to isoform differences or sequencing errors, we have reisolated SS-8 from serum by the established method. Mass spectrometry and peptide analyses are consistent with an N-terminally acetylated form of the Cheng and DeVries sequence (Cheng and DeVries 1991). The verified SS-8 was synthesized and shown to be indistinguishable from the naturally occurring AFP in its cleavage patterns and activity. On the basis of this sequence for SS-8, single alanine residues were substituted by lysine at positions around the helix to sterically block binding of the peptide to ice. Substitutions within the Ala-rich surface inhibited ice binding and lend support to the idea that hydrophobic interactions are involved in binding or partitioning of AFPs to ice. These results conflict with the model implicating lysines in adsorption to ice, but are entirely consistent with the identification of the ice-binding site in type I AFP from winter flounder as the Ala-rich surface.
Results
Re-evaluation of the shorthorn sculpin SS-8 AFP sequence
SS-8 was isolated by the method of Hew et al. (1985) from three different batches of frozen serum collected from different individual fish in early March (2nd and 6th) or early May (6th). In each case, the absorbance and activity profiles generated from the size-exclusion and reversed-phase chromatography steps were as observed previously, and SS-8 eluted from the C-18 column at the appropriate acetonitrile concentration (data not shown). The mass of the purified SS-8 peptide was 3780 daltons. This matched the 42-residue Cheng and DeVries sequence with an N-terminal acetyl group, but was 226 daltons less than the calculated mass (4006 daltons) for the 45-residue sequence deduced by Hew et al. (1985). Endoprotease digestions were done to elucidate this mass discrepancy. Whereas the trypsin digest HPLC profile (Fig. 1 ▶) was essentially the same as that reported previously (Hew et al. 1985), the peptide masses, T1, T2, T3, and T4 matched those for the 42-residue sequence (Fig. 2 ▶) rather than the 45-residue sequence, in which T2 and T4 were discrepant.
Fig. 1.

Reversed-phase HPLC of SS-8 digests. Peptide elution profiles for serum-derived SS-8 are superimposed over those derived from synthetic SS-8. Peptides were detected by A230nm and were designated numerically by order of elution in the acetonitrile gradient (%B). The L, A, and T series refer to digests with endoprotease-Lys-C, endoprotease-Asp-N, and trypsin, respectively.
Fig. 2.

Predicted peptide fragments derived from the putative SS-8 sequence (Cheng and DeVries 1991) and original 45-residue SS-8 sequence (Hew et al. 1985) shown in bold and normal text, respectively. Locations of the predicted peptides generated by endoprotease-Lys-C, endoprotease-Asp-N and trypsin are shown under the sequence. Predicted peptides from the putative sequence are designated as pluses (++++), and from the original sequence are designated as broken lines (—-). The observed masses are tabulated along with the predicted values and sequences from each peptide.
Endoprotease Lys-C digestion of SS-8 was done to circumvent the release of the peptide AAR (residues 10–12), which was too small to be recovered by HPLC. Accordingly, the mass of the AAR segment was conserved in the L4 fragment. In comparing the observed masses of the HPLC peaks for the trypsin and endo-Lys-C digests, both T4 and L4 were 70 daltons more than that of their predicted mass (Fig. 2 ▶). This is consistent with an additional Ala residue in the run between the 14th and 18th residues of the original SS-8 sequence.
Endoprotease Asp-N digestion showed that the second residue was Asp rather than Asn. However, cleavage by endo-Asp-N prior to Asp2 was incomplete and A3 and A2 differed by the mass of an acetylmethionyl residue (173 daltons). A3 also contained a peptide mass (2498 daltons) consistent with partial oxidation of the N-terminal Met. Endo-Asp-N made a second cut after residue 26, to give A1 with a mass of 1314 daltons (Fig. 2 ▶). In the original SS-8 sequence this fragment (A1) would have a mass of 1656. This digestion confirms that the second repeat of KAAA, which is small enough to be lost in tryptic and endo-Lys-C digests, does not exist.
Thus, the evidence from proteolysis and peptide analysis is consistent with the 42-residue sequence being the large serum isoform consistently found in shorthorn sculpin. Determining the correct sequence for this AFP is essential for determining its structure-function relationship and for modeling its interaction with ice. Although the structure of SS-8 has not been determined at atomic resolution, it is known to be largely alpha-helical, and secondary structure predictions suggest that after Pro6 the peptide is one long extended helix.
Delineation of the ice-binding site
We concentrated on mapping the ice-binding site within this region using amino acid substitutions. Initially, Ala to Leu replacement was attempted at several points around the helix to cause steric interference with docking of the peptide to ice. Although this type of substitution was well tolerated in type I AFP from winter flounder (Baardsnes et al. 1999), it caused solubility problems in the shorthorn sculpin AFP (data not shown). Instead, substitutions of Ala to Lys were made to the SS-8 peptide to retain the steric effect of the larger side chain while increasing peptide solubility (Table 1). Several Lys residues were already present in the wild-type peptide, and the addition of a single Lys residue was not expected to have a deleterious effect on the properties and secondary structure of the AFP. Because Lys residues were implicated in the ice-binding mechanism, (Wierzbicki et al. 1996) their use to block ice binding should signify a problem with their location rather than a chemical incompatibility with ice.
Table 1.
Primary sequences of the shorthorn sculpin SS-8 Ala to Lys variants
| 1 | 11 | 21 | 31 | 41 | |
| SS-8 | MDGETPAQKA | ARLAAAAAAL | AAKTAADAAA | KAAAIAAAAA | SA |
| A16K | MDGETPAQKA | ARLAAKAAAL | AAKTAADAAA | KAAAIAAAAA | SA |
| A17K | MDGETPAQKA | ARLAAAKAAL | AAKTAADAAA | KAAAIAAAAA | SA |
| A19K | MDGETPAQKA | ARLAAAAAKL | AAKTAADAAA | KAAAIAAAAA | SA |
| A21K | MDGETPAQKA | ARLAAAAAAL | KAKTAADAAA | KAAAIAAAAA | SA |
| A22K | MDGETPAQKA | ARLAAAAAAL | AKKTAADAAA | KAAAIAAAAA | SA |
| A25K | MDGETPAQKA | ARLAAAAAAL | AAKTKADAAA | KAAAIAAAAA | SA |
Ala to Lys substitutions are shown in bold.
Single Ala to Lys substitutions in the SS-8 peptide at positions 16, 17, 19, 21, 22, and 25 were designed, in turn, to occupy different faces of the α-helix, as can be seen from the end-on view of SS-8 (Fig. 3 ▶). They occurred in the middle of the peptide to maximize the potential disruption to ice binding. Molar ellipticity values for the mutant peptides showed that they have α-helicity equivalent to wild type, although A22K appeared to deviate slightly from optimal helicity. On the basis of the method of Chen et al. (1974), helicity values of the peptides ranged from 69% to 80% (Table 2), within the expected range of the SS-8 isoform (Hew et al. 1980, 1985). Sedimentation equilibrium experiments indicated that the peptides remained predominantly monomeric in solution (Table 2).
Fig. 3.

N-terminal view of SS-8 antifreeze protein from residues 9 to 31. Natural SS-8 Lys residues, K9, K23, and K31 are shown in light blue. (A) The position of A to K substitutions around SS-8. Dark blue Lys side chains (A16K, A19K, and A22K) indicate substitutions that have no effect on antifreeze activity. Red and orange side chains (A17K, A21K, and A25K) indicate severe and moderate effects to antifreeze activity, respectively. A25K (orange) interacts with ice crystals as seen by the bipyramidal morphology but fails to prevent ice crystal growth. (B) Wild-type SS-8 with natural Lys side chains indicated at positions 9, 23, and 31, and the deduced ice-binding plane indicated by the vertical blue line.
Table 2.
Molecular weight determination by sedimentation equilibrium analysis and observed molar elipticity at 1.5°C
| AFP | MW (calculated | MW (observed) ± SD | [Θ] at 222nm at 1.5C (°cm2mol−1) | % Helicity at 1.5°Ca |
| SS-8 | 3780 | 3703 ± 237 | −28850 | 79 |
| A16K | 3837 | 4109 ± 50 | −29780 | 82 |
| A17K | 3837 | 3941 ± 67 | −28380 | 78 |
| A19K | 3837 | 3963 ± 68 | −30230 | 83 |
| A21K | 3837 | 3823 ± 222 | −29280 | 80 |
| A22K | 3837 | 3994 ± 69 | −24980 | 69 |
| A25K | 3837 | 3687 ± 83 | −30080 | 82 |
a Helicity calculations are based on the method of Chen et al. (1974) using k = 4.6 and [θ]∞ = −41,000 deg.cm2/dmole (Gans et al. 1991).
The single substitutions A16K, A19K, and A22K on the hydrophilic surface of SS-8 did not affect antifreeze activity. Stock solutions of these variants were assayed over a wide range of concentrations (0–8 mg/mL) and showed no major deviation from wild-type thermal hysteresis values, although the values for A22K were slightly suboptimal (Fig. 4 ▶), nor did they show any alteration in their elongated hexagonal bipyramidal crystal morphology or a tendency for these crystals to grow during the thermal hysteresis measurements (Fig. 5 ▶).
Fig. 4.

Thermal hysteresis activity as a function of antifreeze protein concentration (mg/mL). Active antifreeze variants are: WT (○), A16K (○), A19K (▾), A22K (▿). Inactive antifreeze variants are: A17K (▪), A21K (□), and A25K (♦). Each data point represents the mean of three determinations and the vertical bars represent the standard deviation.
Fig. 5.

Ice crystal morphology in the presence of wild-type SS-8 and variants. (A) Ice crystals formed in the presence of 1.0 mg/mL WT, A16K, A19K, A22K, or A25K in 0.1 M NH4HCO3. The samples were undercooled to 0.09°C over 75 sec, after which image collection timing began from time zero. SS-8 variants that generated wild-type ice crystal morphology included A16K, A19K, and A22K. The inactive variant A25K ice crystal grew slowly over the 10-min interval of measurement, and generated an elongated hexagonal bipyramid. (B) Ice crystals of the highly inactive samples were formed in the presence of 1.0 mg/mL A17K or A21K in 0.1 M NH4HCO3, and image collection began immediately after ice crystal stabilization at 0.02°C of undercooling.
In contrast, single substitutions A17K, A21K, and A25K on the hydrophobic surface of SS-8 abolished all AFP activity in the mutant peptides (Fig. 4 ▶). The A17K and A21K substitutions were particularly severe because they could not generate ice bipyramids at protein concentrations below 4 mg/mL (Fig. 5 ▶). Above 4 mg/mL, ice bipyramids were formed, but they grew rapidly during thermal hysteresis measurements (data not shown). Lysine substitution at A25 had less severe effects than at A17 and A21. A25K produced bipyramidal ice crystals, but these showed slow growth over the entire concentration range tested, indicating that the peptide still had significant interactions with the ice surface but could not prevent the addition of water to the ice lattice. Therefore, by definition, this mutation abolished AFP activity. Because A25K was on the borderline of activity, it helped delineate the ice-binding face of SS-8 as being that section of the Ala-rich surface adjacent to the hydrophilic face and not just any section of the hydrophobic side. The complete absence of shaping of the A17K ice crystal at 1 mg/mL (Fig. 4 ▶) indicates that this is the most crucial substitution on the Ala-rich face.
SS-8 N-terminal cap structure
Molecular dynamics simulations of the initially totally α-helical SS-8 peptide resulted in the formation of an N-cap structure, coinciding with a favorable decrease in energy of ∼80 kcal/mole (Fig. 6A ▶). The N-cap structure has two predominant salt bridges, between residues Asp 2/Lys 9 and Glu 4/Arg 12, with other intramolecular hydrogen bonds forming between Gln 8/Arg 12 and Asp 2/Thr 5. The hydrophobic Met 1 side chain extends into the cleft formed between Leu 13 and Ala 16, whereas a turn exists between residues Thr 5 to Gln 8. This cap structure folds the helix N terminus up and away from the Ala-rich surface such that it does not sterically interfere with the ice-binding face.
Fig. 6.
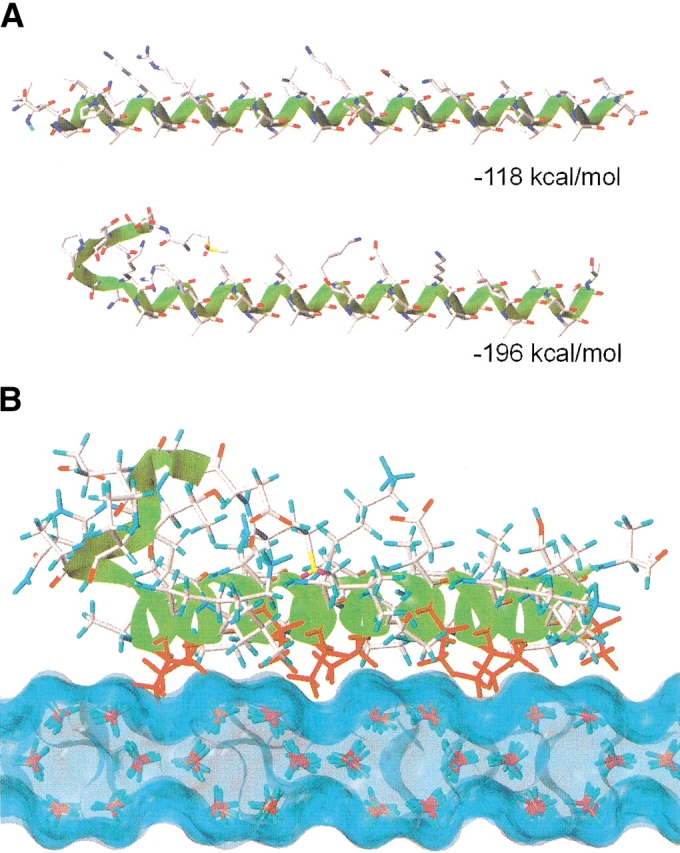
Model of SS-8 and interaction with the secondary prism plane of ice. (A) N-terminal cap structure of SS-8. The all α-helix model of SS-8 (top) (Wierzbicki et al. 1996), compared with the new low energy N-terminal cap structure generated from molecular dynamics simulation (bottom). Total energy as determined by energy minimization for each conformer is indicated. (B) SS-8 docking to the secondary prism plane of ice. The N terminus is shown on the left and the green ribbon traces the backbone. Ala residues within the ice-binding plane delineated by Ala 17 and Ala 21 are colored red. Note how the Ala side chains that repeat at 11 -amino acid intervals fit into the grooves of the ice surface. The Connolly ice surface shown was generated by SYBYL 6.6.
Modeling of SS-8 on the secondary prism plane
The optimal modeled solution found with our automated docking procedure resulted in the SS-8 peptide aligned across the secondary prism face of ice with the helical axis parallel to the <−1102> direction, similar to the helix alignment found by Wierzbicki et al. (1996) and Madura et al. (2000). In contrast, however, our model showed a preference for the alanine-rich surface contacting the ice rather than the lysine-rich side. Alanine residues 10, 14, 21, 25, 32, and 36 have surface complementarity with the secondary prism face grooves (Fig. 6B ▶). In our model, the lysine chains are directed away from the ice into the aqueous phase, where they are free to move and interact with liquid water. Docking of the all α-helix model (Wierzbicki et al. 1996) to the same ice surface also showed favorable interactions via the conserved Ala face (data not shown), indicating that adsorption via the Ala face is not influenced by the modeled cap structure.
The docking simulations did not use the position of the inactivating Lys substitutions as parameters for determination of the best AFP–ice interaction. Therefore, the activity data provided by the variant peptides provide positive verification that the correct protein–ice interaction was determined. Ala 17 and Ala 21 are oriented so that the side chains are almost perpendicular to the ice surface; hence, substitutions with large Lys residues would generate a severe steric clash with the ice surface.
Discussion
The mass difference between the two versions of the SS-8 sequence can be accounted for by three changes as follows: (1) replacement of Asn 2 by Asp; (2) insertion of an additional Ala in the first long Ala-tract; and (3) deletion of the KAAA duplication. Although it is possible that the original 45-residue sequence (Hew et al. 1985) represents a rare SS-8 isoform, it seems very unlikely now, given that three preparations of SS-8 purified from different fish on separate occasions by the established protocol showed the same 226-dalton mass discrepancy. Moreover, the sequence reported for the analogous AFP isoform in the serum of the closely related grubby sculpin (Chakrabartty and Hew 1988) has all three of the above changes and is only missing the last two residues. Automated Edman degradation done in spinning cup sequencers typically missed the last few residues in a sequence and it is likely that the 40-residue sequences reported for the shorthorn and grubby sculpins (Chakrabartty and Hew 1988; Knight et al. 1991) are both longer by the C-terminal SA residues.
It is important to establish that there is only one SS-8 sequence, because the compilation of isoform diversity can be used to identify those features of the structure that are highly conserved. Comparison of five different type I AFP sequences within righteye flounders showed that the Ala-rich surface was far better conserved than the repeating Thr/Asx face, which had been suggested previously as the ice-binding surface (DeVries 1983; Sicheri and Yang 1995). Moreover, substitution experiments that replaced Thr with Val weakened the notion of ice binding via hydrogen-bond formation (Chao et al. 1997; Haymet et al. 1998; Zhang and Laursen 1998). This led to a re-evaluation of the ice-binding surface by Baardsnes et al. (1999) with the realization that the Ala-rich surface adjacent to the Thr repeats is the region of primary contact with ice. In turn, this has also led us to question the involvement of the Lys-rich hydrophilic helix face of SS-8 in ice binding. Our substitution experiments suggest that Lys residues at positions 9, 23, and 31, and Arg 12 do not directly interact with the ice surface. If the AFP binds to the ice surface via these basic residues, substitutions at positions 16 and 19 would disrupt the putative 11 residue spacing (16.9Å) between Arg 12 and Lys 23, and the 33.8Å spacing between Lys 9 and Lys 31. The spacing between these residues is a key feature of the model by Wierzbicki et al. (1996) and yet the appearance of an additional lysyl side chain at either Ala 16 or Ala 19 had no adverse effect on antifreeze activity or ice crystal morphology. Previously, the loss of antifreeze activity on modification of Lys residues with fluorescein has been used as support of the Lys-binding mechanism (DeVries 1982). However, no substantiating evidence regarding the helicity and solubility of these peptides has been presented. If A16K and A19K are well tolerated on the putative ice-binding face of the α-helix, it is hard to explain why the A17K, A21K, and A25K substitutions are so disruptive on the opposite side of the helix.
On the basis of our interpretation of the amino acid substitutions reported here, the ice-binding site of SS-8 is the Ala-rich surface immediately adjacent to the hydrophilic surface. This surface is exactly analogous to the redefined ice-binding site of the flounder type I AFPs, where the junction with the hydrophilic surface is marked by the repeating Thr residues at 11 amino acid intervals (T2, T13, T24, T35). A leucine, a threonine, and an isoleucine occupy equivalent positions in SS-8 (Fig. 7 ▶). It would be interesting to know whether the structural diversity in this location affects the binding specificity of SS-8 and directs it to bind to the secondary prism planes rather than the pyramidal planes (Knight et al. 1991; Laursen et al. 1994). Haymet et al. (1999), in a related experiment, replaced the repeating Thr of the flounder AFP with Ala. This caused a substantial loss of antifreeze activity and also altered the binding pattern of the AFP to ice from flounder-like to sculpin-like.
Fig. 7.

Helical net representations of natural and synthetic type I peptides. The natural shorthorn sculpin SS-8 and winter-flounder HPLC-6 isoforms are shown along with the synthetic peptides AKAAK, LKAAK, poly-AK (Zhang and Laursen 1999), and 43-mer (Wierzbicki et al. 2000). Red residues indicate positions of inactivating steric mutations, green represent positions unaffected by these mutations (ibid; Baardsnes et al. 1999). Thr and Lys residues are shown in light pink and light blue, respectively. The putative Ala-rich binding face is indicated by the dark-blue rectangle.
Judging by the full activity of A19K and A22K, the appearance of a lysyl side chain just anywhere within the Ala-rich surface is not sufficient to disrupt ice adsorption and, therefore, the effect is stereo specific and not simply due to the insertion of a hydrophilic group in a hydrophobic surface. In this regard, Ala 22 lies well within the Ala-rich surface, which spans two thirds of the way around the helix (Fig. 4 ▶). Ala 19 defines a junction between the Ala-rich and hydrophilic surfaces, almost diametrically opposite to Ala 17 where lysine insertion eliminates antifreeze activity. The junction at Ala 19 is not a part of the ice-binding site, but the junction at Ala 17 clearly is. The complete loss of activity and ability of this variant to shape ice crystals below 4.0 mg/mL (Fig. 5B ▶) indicates that this is the most crucial position on the Ala-rich surface for ice-binding. It would appear to be analogous to Ala 17 of the HPLC-6 winter-flounder isoform that lies adjacent to the position occupied by the conserved Thr residues.
A useful check of our hypothesis is provided by a series of synthetic antifreeze peptides made by Zhang and Laursen (1999), and Wierzbicki et al. (2000) (Fig. 7 ▶). These α-helical peptides were based loosely on the winter-flounder AFPs and contained Ala and Lys as the principle amino acids. The peptides (AKAAK, LKAAK, and the 43-mer) with regularly spaced lysines on one side of the helix showed weak thermal hysteresis activity and shaping of ice crystals. The peptides with irregularly spaced lysines were devoid of activity (poly-AK). The interpretation placed on these results by the authors was that the lysines, when ideally spaced by the helix backbone, function in binding the peptide to ice. We contend that they unintentionally provided an Ala-rich surface flanking a hydrophilic face, which is the structural feature required for ice binding that the flounder and sculpin type I AFPs have in common. As can be seen in Figure 7 ▶, an uninterrupted Ala-rich face is present in shorthorn sculpin SS-8 and the winter flounder HPLC-6 isoform, and those three synthetic peptides that have ice-binding activity. The peptides generated by Zhang and Laursen (1999), and Wierzbicki et al. (2000), had very low activity compared with wild-type SS-8. This is possibly due to the lack of a stabilizing N-terminal cap structure and the absence of residues that may duplicate the function of L13, T24, and I35 on the periphery of the ice-binding face of SS-8. Predictably, the irregular placement of lysines around the helix of peptide poly-AK (Fig. 7 ▶) disrupts the Ala-rich surface in the same way that A17K, A21K, and A25K inhibit SS-8 activity, and A17L and A21L substitutions disrupt the winter-flounder HPLC-6 activity (Baardsnes et al. 1999).
Ice-docking simulations of SS-8 with the secondary prism plane were performed to complement our biochemical evidence for the new ice-binding surface. One interesting outcome of molecular dynamic simulations was the generation of a N-terminal cap structure that stabilizes the peptide by ∼80 kcal/mole over the all α-helix model through the formation of several intramolecular hydrogen bonds (Fig. 6A ▶). This cap structure is initiated around Pro 6, a known helix breaker (Chou and Fasman 1978), and could account for the 20% reduction in helicity of SS-8 compared with winter-flounder HPLC-6. Previous docking of SS-8 to ice used an all α-helix model (Wierzbicki et al. 1996; Madura et al. 2000). We performed solvated computer simulation of SS-8 on the {11–20} plane of ice in order to evaluate three possible docking models. The best docking solution of SS-8 on the {11–20} plane of ice placed the Ala-rich surface in contact with the ice, agreeing independently with our biochemical evidence. In addition, the helix axis was aligned along the <−1102> direction. Overall, the total energy of the best docking, mirror-image docking in the <1–102> direction, and a docking of the Lys-containing surface aligned along the <−1102> direction were −820.2, −788.3, and −762.1 kcal/mole, respectively. The energetic gain between the best docking and mirror-related docking was mainly due to favorable van der Waals interactions, in which the threads of the helix form a highly complementary fit to the grooves of the ice surface (Fig. 6B ▶). Comparing the best docking with the lysine face docking, there is a small 9 kal/mole gain from van der Waals interactions; however, most of the gain is realized in the electrostatic contributions (49 kcal/mole). This result suggests that it is more energetically favorable to have the lysines facing away from the ice because the Lys (and Arg) residues will then be able to adopt ideal hydrogen-bonding conformations with solvating water molecules (Sönnichsen et al. 1996; Harding et al. 1999).
These results indicate that the mode of binding of all of the type I isoforms works via the conserved Ala-rich face. The Lys residues likely act to solubilize the peptide to facilitate the Ala-face interaction with the ice surface. These results are important to generate the correct ice–protein interface for computer simulations that are crucial for modeling ice–protein interactions.
Materials and methods
Purification of shorthorn sculpin SS-8 AFP
Shorthorn sculpin serum was thawed and clarified by centrifugation prior to fractionation on a Sephadex G-75 size exclusion column (16 cm × 100 cm). Fractions (4 mL) were eluted in 0.1M NH4HCO3 buffer (pH 7.9) at a flow rate of 0.5 mL/min according to the method of Hew et al. (1985) and were assayed for thermal hysteresis activity (Chakrabartty and Hew 1991). Active fractions were pooled, lyophilized, dissolved in 0.1% TFA, and rechromatographed by reversed-phase HPLC on a Vydac C-18 semipreparative column (1.0 cm × 25 cm) with an 80% acetonitrile/0.1% TFA gradient of 0% to 100% in 100 min at a flow rate of 3 mL/min. SS-8 eluted at 61% acetonitrile and was characterized by mass spectrometry and proteolysis.
Enzymatic digests
Native SS-8 (100 μg) or synthetic peptide (80 μg) was digested with trypsin (Sigma), sequencing grade endoprotease Lys-C (BMC) or sequencing grade endoprotease Asp-N (BMC). Trypsin digests were done in 0.1 M NH4HCO3 (pH 8.0), 0.1 M CaCl2 at 37°C in a total volume of 200 μL with an enzyme:substrate ratio of 1:20 (w/w). After 1 h, an additional aliquot of enzyme (10 μg) was added and the digest continued for another hour. Endo Lys-C digests were performed in 25 mM Tris-HCl (pH 8.0), 1 mM EDTA at an enzyme/substrate ratio of 1:125 (w/w) in a total volume of 200 μL and incubated for 3 h at 37°C. Endo Asp-N digests were done in 20 mM Tris-HCl (pH 8.0) at a 1:100 enzyme:substrate ratio in a total volume of 400 μL with incubation for 3 h at 37°C. Digestion products were separated on an analytical reversed-phase C-18 Vydac column (0.5 cm × 25 cm) using an 80% acetonitrile/0.1% TFA gradient from 0% to 60% in 60 min.
Mass analysis
The masses of sculpin AFP (native and synthetic variants) and their proteolytic fragments were determined on a Kratos Analytical MALDI Mass Spectrometer and/or on a Fisons VG Quattro triple quadropole mass spectrometer (Manchester) fitted with an electrospray ionization source (Houston et al. 1998).
Thermal hysteresis activity and photomicroscopy
Thermal hysteresis activity was measured using a nanoliter osmometer (Clifton Technical Physics) as described by Chakrabartty and Hew (1991). Thermal hysteresis is defined as the temperature difference (°C) between the melting point and the nonequilibrium freezing point of a solution. Ice growth of >0.2 μm/sec signifies that the solution freezing point has been reached or exceeded. Ice crystal morphology was observed using a Leitz 22 microscope and recorded by a Panasonic CCTV camera linked to a JVC Super VHS video recorder. Still images were obtained from a Silicon Graphics INDY terminal using IRIS Capture version 1.2.
Peptide synthesis and quantification
SS-8 and its variants were made at PENCE (University of Alberta) by solid-phase peptide synthesis as described previously (Hodges et al. 1988; Gronwald et al. 1996). Peptide concentrations were determined by amino acid analysis. Samples were hydrolyzed (6N HCl at 160°C for 1.5 h) and analyzed in a Beckman Model 6300 amino acid analyzer (San Ramon). Norleucine was used as an internal reference to correct the amount of each identified amino acid.
Circular dichroism spectroscopy
CD spectra were collected as described previously (Chao et al. 1996) using a Jasco J-500C spectropolarimeter (Jasco). The helicity of the protein was monitored at 222 nm during temperature denaturation studies. Each data point was the average of a minimum of 24 readings. The buffer used was 50 mM potassium phosphate (pH 7.0), in 50 mM KCl.
Sedimentation equilibrium ultracentrifugation
Sedimentation equilibrium experiments were carried out at 5°C in a Beckman XL-1 analytical ultracentrifuge using absorbance optics following the procedures described by Laue and Stafford (1999). Aliquots (110 μL) of sample solution were loaded into 6-sector CFE sample cells, allowing three concentrations of sample to be run simultaneously. Runs were performed at a minimum of two different speeds and each speed was maintained until there was no significant difference in r2/2 versus absorbance scans taken 2-hours apart to ensure that equilibrium was achieved.
The sedimentation equilibrium data was evaluated using the NONLIN program, which incorporates a nonlinear least-squares curve-fitting algorithm described by Johnson et al. (1981). This program allows the analysis of both single and multiple data files. Data can be fit to either a single ideal species model or models containing up to four associating species, depending on which parameters are permitted to vary during the fitting routine. The protein's partial specific volume and solvent density were estimated using the SEDNTERP program, which incorporates calculations detailed by Laue et al. (1991).
Peptide modeling
The SS-8 isoform model was constructed using SYBYL 6.6 (Tripos Associates) on the basis of standard α-helical geometry and the 42 amino acid sequence: Ac-MDGETPAQKAARLAAAAAA LAAKTAADAAAKAAAIAAAAASA. Energy minimization and dynamics simulations were carried out using the Amber 4.1 force field with Kollman All-atom charges and a constant dielectric. Only the TIP3 water model was used in simulations for both the aqueous phase and the ice (Jorgensen et al. 1983). Minimization was gradient terminated at 0.05 kcal/mole. An initial gentle dynamic simulation was performed on the minimized structure without solvent to reduce computational time for a length of 300 pico-seconds (ps) at a temperature of 150 K. A low-energy conformer derived from the dynamics, with a new N terminus cap structure, was then subjected to further minimization before being used for a docking study. A secondary prism face of ice (∼36 × 80 × 5Å), was generated using SYBYL, with the oxygen atoms aggregated to maintain ice integrity throughout the simulations and was subjected to a brief dynamics and minimization routine to randomize the positions of the hydrogen atoms of ice. A docking algorithm, written using the SYBYL Programming Language systematically docked the peptide to the ice. A full 360 degrees of rotational freedom were sampled both parallel to the helix axis and perpendicular to the axis of the peptide on the plane of the ice. In addition, the peptide had full translational freedom across the ice surface.
The best-docked solution from the docking procedure, (with the peptide aligned along the <−1 1 0 2> direction and the alanine face toward the ice), was then trimmed and solvated using periodic boundary conditions to simulate the binding of the peptide at the aqueous interface (∼80 × 35 × 40Å solvation box). This model was again minimized to completion using the previous parameters.
Two other orientations of the SS-8 peptide bound to ice were investigated for comparison, the mirror image of the best-docked orientation (with the peptide aligned along the <1 −1 0 2> direction), and also with the lysine-rich face directed toward the ice in the original <−1 1 0 2> orientation. After these solvated models were minimized, the energy of interaction with each phase (ice and water) was calculated. This procedure involved separating the peptide, solvent, and ice into separate molecular files and then calculating the interaction energy of the peptide with each phase by measuring the difference in energy when the peptide was in its original position with respect to the phase and when it was moved ∼50Å away from the phase. These energy differences could be further resolved into electrostatic and van der Waals energy contributions.
Acknowledgments
PENCE and A/F Protein, Inc. funded this work. We are grateful to Garth Fletcher for the gift of shorthorn sculpin serum, Marc Genest and Paul Semchuk for peptide synthesis, Isabel Werner for CD measurements, and Leslie Hicks for sedimentation equilibrium analyses. We thank Sherry Gauthier and Blair Besley for reevaluation of the SS-8 sequence.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.26501.
References
- Baardsnes, J., Kondejewski, L.H., Hodges, R.S., Chao, H., Kay, C., and Davies, P.L. 1999. New ice-binding face for type I antifreeze protein. FEBS Lett. 463 87–91. [DOI] [PubMed] [Google Scholar]
- Chakrabartty, A. and Hew, C.L. 1988. Primary structures of the alanine-rich antifreeze polypeptides from grubby sculpin, Myoxocephalus aenaeus. Can. J. Zool. 66 403–408. [Google Scholar]
- Chakrabartty, A. and Hew, C.L.1991. The effect of enhanced alpha-helicity on the activity of a winter flounder antifreeze polypeptide. Eur. J. Biochem. 202 1057–1063. [DOI] [PubMed] [Google Scholar]
- Chao, H., Hodges, R.S., Kay, C.M., Gauthier, S.Y., and Davies, P.L. 1996. A natural variant of type I antifreeze protein with four ice-binding repeats is a particularly potent antifreeze. Protein Sci. 5 1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, H., Houston, Jr., M.E., Hodges, R.S., Kay, C.M., Sykes, B.D., Loewen, M.C., Davies, P.L., and Sönnichsen, F.D. 1997. A diminished role for hydrogen bonds in antifreeze protein binding to ice. Biochemistry 36 14652–14660. [DOI] [PubMed] [Google Scholar]
- Chen, Y.H., Yang, J.T., and Chau, K.H. 1974. Determination of the helix and b form of proteins in aqueous solution by circular dichroism. Biochemistry 13 3350–3359. [DOI] [PubMed] [Google Scholar]
- Cheng, C.H. and DeVries, A.L. 1991. The role of antifreeze glycopeptides and peptides in the freezing avoidance of cold-water fish. In Life under extreme conditions. pp. 1–14. Springer-Verlag. Berlin, Germany
- Chou, K.C. 1992. Energy-optimized structure of antifreeze protein and its binding mechanism. J. Mol. Biol. 223 509–517. [DOI] [PubMed] [Google Scholar]
- Chou, P.Y. and Fasman, G.D. 1978. Prediction of the secondary structure of proteins from their amino acid sequence. Advan. Enzymol. 47 45–148. [DOI] [PubMed] [Google Scholar]
- Dalal, P. and Sönnichsen, F.D. 2000. Source of the ice-binding specificity of antifreeze protein type I. J. Chem. Inf. Comput. Sci. 40 1276–1284. [DOI] [PubMed] [Google Scholar]
- Deng, G., Andrews, D.W., and Laursen, R.A. 1997. Amino acid sequence of a new type of antifreeze protein, from the longhorn sculpin Myoxocephalus octodecimspinosis. FEBS Lett. 402 17–20. [DOI] [PubMed] [Google Scholar]
- Devries, A.L. 1982. Biological antifreeze agents in coldwater fishes. Comp. Biochem. Physiol. 73A 627–640. [Google Scholar]
- Devries, A.L. 1983. Antifreeze peptides and glycopeptides in cold-water fishes. Annu. Rev. Physiol. 45 245–260. [DOI] [PubMed] [Google Scholar]
- DeVries, A.L. and Lin, Y. 1977. Structure of a peptide antifreeze and mechanism of adsorption to ice. Biochim. Biophys. Acta. 495 388–392. [DOI] [PubMed] [Google Scholar]
- Ewart, K.V., Rubinsky, B., and Fletcher, G.L. 1992. Structural and functional similarity between fish antifreeze proteins and calcium-dependent lectins. Biochem. Biophys. Res. Commun. 185 335–340. [DOI] [PubMed] [Google Scholar]
- Fletcher, G.L, Hew, C., and Davies P.L. 2001. Antifreeze proteins of teleost fishes. Annu. Rev. Physiol. 63 359–390. [DOI] [PubMed] [Google Scholar]
- Gans, P.J., Lyu, P.C., Manning, M.C., Woody, R.W., and Kallenbach, N.R. 1991. Biopolymers 31 1605–1614 [DOI] [PubMed] [Google Scholar]
- Gronwald, W., Chao, H., Reddy, D.V., Davies, P.L., Sykes, B.D., and Sönnichsen, F.D. 1996. NMR characterization of side chain flexibility and backbone structure in the type I antifreeze protein at near freezing temperatures. Biochemistry 35 16698–16704. [DOI] [PubMed] [Google Scholar]
- Harding, M.M., Ward, L.G., and Haymet, A.D. 1999. Type I `antifreeze' proteins. Structure-activity studies and mechanisms of ice growth inhibition. Eur. J. Biochem. 264 653–665. [DOI] [PubMed] [Google Scholar]
- Haymet, A.D., Ward, L.G., Harding, M.M., and Knight, C.A. 1998. Valine substituted winter flounder `antifreeze': Preservation of ice growth hysteresis. FEBS Lett. 430 301–306. [DOI] [PubMed] [Google Scholar]
- Haymet, A.D., Leanne, G.W., and Harding, M.M. 1999. Winter flounder "antifreeze" proteins: Synthesis and ice growth inhibition of analogues that probes the relative importance of hydrophobic and hydrogen-bonding interactions. J. Am. Chem. Soc. 121 941–948. [Google Scholar]
- Hew, C.L., Fletcher, G.L., and Ananthanarayanan, V.S. 1980. Antifreeze proteins from the shorthorn sculpin, Myoxocephalus scorpius: Isolation and characterization. Can. J. Biochem. 58 377–383. [DOI] [PubMed] [Google Scholar]
- Hew, C.L., Joshi, S., Wang, N.C., Kao, M.H., and Ananthanarayanan, V.S. 1985. Structures of shorthorn sculpin antifreeze polypeptides. Eur. J. Biochem. 151 167–172. [DOI] [PubMed] [Google Scholar]
- Hodges, R.S., Semchuk, P.D., Taneja, A.K., Kay, C.M., Parker, J.M.R., and Mant, C.T. 1988. Protein design using model synthetic peptides. Pept. Res. 1 19–30. [PubMed] [Google Scholar]
- Houston, Jr., M.E., Chao, H., Hodges, R.S., Sykes, B.D., Kay, C.M., Sönnichsen, F.D., Loewen, M.C., and Davies, P.L. 1998. Binding of an oligopeptide to a specific plane of ice. J. Biol. Chem. 273 11714–11718. [DOI] [PubMed] [Google Scholar]
- Johnson, M.L., Correia, J.J., Yphantis, D.A., and Halvorson, H.R. 1981. Analysis of data from the analytical ultracentrifuge by non-linear least-squares techniques. Biophys. J. 36 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, W.L., Chandrasekar, J., Madura, J.D., Impey, R.W., and Klein, M.L. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 926–935. [Google Scholar]
- Knight, C.A., Cheng, C.C., and DeVries, A.L. 1991. Adsorption of alpha-helical antifreeze peptides on specific ice crystal surface planes. Biophys. J. 59 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laue, T.M. and Stafford, III, W.F. 1999. Modern applications of analytical ultracentrifugation. Annu. Rev. Biophys. Biomol. Struct. 28 75–100. [DOI] [PubMed] [Google Scholar]
- Laue, T.M., Shah, B.D., Ridgeway, T.M., and Pelletier, S.L. 1991. Computer-aided interpretation of analytical sedimentation data for proteins. In Analytical ultracentrifugation in biochemistry and polymer science. (ed. S.E. Harding, A.J. Rowe, and J.C. Horton). Royal Society of Chemistry, Cambridge, UK.
- Laursen, R.A., Wen, D., and Knight, C.A. 1994. Enantioselective adsorption of the D- and L- forms of an alpha-helical antifreeze polypeptide to the {20–21} planes of ice. J Amer. Chem. Soc. 116 12057–12058. [Google Scholar]
- Loewen, M.C., Chao, H., Houston, Jr., M.E., Baardsnes, J., Hodges, R.S., Kay, C.M., Sykes, B.D., Sönnichsen, F.D., and Davies, P.L. 1999. Alternative roles for putative ice-binding residues in type I antifreeze protein. Biochemistry 38 4743–4749. [DOI] [PubMed] [Google Scholar]
- Madura, J.D., Baran, K., and Wierzbicki, A. 2000. Molecular recognition and binding of thermal hysteresis proteins to ice. J. Mol. Recognit. 13 101–113. [DOI] [PubMed] [Google Scholar]
- Raymond, J.A. and DeVries, A.L. 1977. Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc. Natl. Acad. Sci. 74 2589–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri, F. and Yang, D.S. 1995. Ice-binding structure and mechanism of an antifreeze protein from winter flounder. Nature 375 427–431. [DOI] [PubMed] [Google Scholar]
- Sönnichsen, F.D., DeLuca, C.I., Davies, P.L., and Sykes, B.D. 1996. Refined solution structure of type III antifreeze protein: Hydrophobic groups may be involved in the energetics of the protein-ice interaction. Structure 41325–37. [DOI] [PubMed] [Google Scholar]
- Wen, D. and Laursen, R.A. 1992. A model for binding of an antifreeze polypeptide to ice. Biophys. J. 63 1659–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki, A., Taylor, M.S., Knight, C.A., Madura, J.D., Harrington, J.P., and Sikes, C.S. 1996. Analysis of shorthorn sculpin antifreeze protein stereospecific binding to (2–10) faces of ice. Biophys. J. 71 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki, A., Knight, C.A., Rutland, T.J., Muccio, D.D., Pybus, B.S., and Sikes, C.S. 2000. Structure-function relationship in the antifreeze activity of synthetic alanine-lysine antifreeze polypeptides. Biomacromolecules 1 268–274. [DOI] [PubMed] [Google Scholar]
- Yang, D.S., Sax, M., Chakrabartty, A., and Hew, C.L. 1988. Crystal structure of an antifreeze polypeptide and its mechanistic implications. Nature 333 232–237. [DOI] [PubMed] [Google Scholar]
- Zhang, W. and Laursen, R.A. 1998. Structure-function relationships in a type I antifreeze polypeptide. The role of threonine methyl and hydroxyl groups in antifreeze activity. J. Biol. Chem. 273 34806–34812. [DOI] [PubMed] [Google Scholar]
- Zhang, W. and Laursen, R.A. 1999. Artificial antifreeze polypeptides: alpha-helical peptides with KAAK motifs have antifreeze and ice crystal morphology modifying properties. FEBS Lett. 455 372–376. [DOI] [PubMed] [Google Scholar]