

Abstract
It has been shown previously that some membrane proteins have a conserved core of amino acid residues. This idea not only serves to orient helices during model building exercises but may also provide insight into the structural role of residues mediating helix–helix interactions. Using experimentally determined high-resolution structures of α-helical transmembrane proteins we show that, of the residues within the hydrophobic transmembrane spans, the residues at lipid and subunit interfaces are more evolutionarily variable than those within the lipid-inaccessible core of a polypeptide's transmembrane domain. This supports the idea that helix–helix interactions within the same polypeptide chain and those at the interface between different polypeptide chains may arise in distinct ways. To show this, we use a new method to estimate the substitution rate of an amino acid residue given an alignment and phylogenetic tree of closely related proteins. This method gives better sensitivity in the otherwise-conserved transmembrane domains than a conventional similarity analysis and is relatively insensitive to the sequences used.
Keywords: Protein structure, lipid bilayer, evolutionary conservation, sequence alignment, phylogeny
Membrane proteins with at least one transmembrane α-helix account for >25% of the proteins in almost every genome sequenced so far (Stevens and Arkin 2000). They are also the therapeutic targets for most of the drugs currently in medicinal use. However, as the native structures of membrane proteins require the presence of a lipid bilayer or substitute amphiphile, experimental determination of structures is extremely difficult. Thus, α-helical transmembrane proteins are severely under-represented in protein structural databases (Sakai and Tsukihara 1998). Of all the (non-homologous) proteins in the Protein Data Bank (PDB) (Bernstein et al. 1977) there about a dozen high resolution (<3 Å) α-helical transmembrane protein structures.
With this set of structures we can begin trying to understand why the membrane domain folds the way it does. This important class of proteins exists in an environment quite unlike that of aqueous proteins. Thus, the folding membrane domain has a different set of guiding influences that govern the final form. For example, without a polar solvent the bilayer-bound peptide backbone almost always fulfills its hydrogen bonding potential with an α-helical secondary structure. The two-stage model of membrane protein folding and oligomerization (Popot and Engelman 1990) has been used as a basis for understanding transmembrane domain formation. This model considers the thermodynamic motivation for helices to fold and then to associate laterally within the bilayer. By investigating the patterns of membrane domain structure we can build on this basic framework to show how a particular association of helices generates the final structure. In particular, the two-stage model does not distinguish mechanistically between helices from the same or different polypeptide chains.
The structural characteristics of an α-helical bundle make membrane domains worthy targets for molecular modeling, compensating for the deficit in high resolution structures. However, molecular modeling of transmembrane helical bundles often results in multiple low-energy structures. These require additional low-resolution structural information to distinguish between false minima and the native protein structure. This augmentation can come in the form of empirical data (Arkin et al. 1997) or from constraints based on knowledge of the biological function and folding. Such constraints include the placing of biologically functional residues in appropriate positions and locating substitution-sensitive residues in particular positions, most notably at helix–helix interfaces (Treutlein et al. 1992; Adams et al. 1995, 1996). Consistent with this approach, previous studies have looked at evolutionarily conserved residues and placed them in a structural context (Donnelly et al. 1993). These analyses have formed the basis of the principle that membrane proteins have conserved cores. However, the initial observations of a conserved transmembrane core were limited to the analysis of single proteins. Hence, the deduction may be subject to the peculiarities of the particular protein chosen. For example, when considering hydrophobicity, significant proportions of hydrophilic residues in a transmembrane protein core seem specific to a few proteins (e.g., rhodopsins).
Using the recently increased number of high resolution structures of α-helical membrane domains, we have examined a database of non-homologous hydrophobic α-helical bundles. We present a rigorous and statistical approach for exploring the structural significance of evolutionary variation in transmembrane α-helix residues. Not only do we analyze a much larger data set than prior analyses, but we also make use of a new technique for calculating the evolutionary preservation of protein residues based on estimated substitution rates (rather than residue similarity). This technique is intentionally insensitive to the choice of related sequences used to generate conservation data. Our results show that the cores of helical transmembrane proteins, as a class, are indeed more conserved than the lipid-exposed regions. However, this conservation seems to be strongest at the core of individual polypeptide chains and possibly biologically active homo-oligomers thereof. As a class, the oligomerizing surfaces within the transmembrane domains are not well-conserved and in this respect are similar to the lipid-facing surfaces.
Results
Substitution rate of accessible residues
The distribution of relative substitution rates for three different residue classes is shown in Figure 1 ▶. Buried residues are defined as those that have <7% of the maximum solvent-accessible surface area (Hubbard and Blundell 1987) exposed to a solvent probe (1.4 Å diameter). The distribution of substitution rate indices for buried residues is biased towards lower values compared to both the solvent-exposed residues (given the native structure) and the oligomer-interface residues. The differences are particularly apparent for Ri values >1.5; there are approximately twice as many exposed residues than buried ones. The mean vales of Ri are 0.88, 1.09, and 1.00 for the buried, exposed, and oligomer-interface classes, respectively. When the accessibility calculation uses single chains, rather than oligomeric structures, the distribution of relative rates is virtually unaltered (not shown). In effect, by using the single-chain accessibility, the oligomer-interface residues are reclassified as solvent-exposed. With a distribution similar to the solvent-exposed residues, this reclassification makes little difference.
Fig. 1.

The distribution of substitution rate indices Ri for buried, exposed, and oligomer-interface transmembrane residues. A substitution rate index >0 indicates a substitution rate higher than the alignment average.
Protein pictures
Figure 2 ▶, a representation of bacteriorhodopsin from the structural database, illustrates a clear instance in which the most variable residues are found around the periphery of the protein and the most preserved are in the center. The picture of a bacteriorhodopsin subunit shows a more variable exterior, with no clear distinction between lipid-contacting surfaces and oligomerizing surfaces. Also, this illustrates that the use of estimated substitution rates gives a picture with better gradation than is generated from conservation values. Figure 3 ▶ shows how the Ri of residues can be used to generate vectors that illustrate which side of the transmembrane helices are most preserved. For large hetero-oligomeric protein complexes, such as cytochrome c oxidase, coloring individual residues according to conservation or substitution rate (Fig. 3A ▶) does not produce such a clear correlation, although it may noted that the variable residues seem to be around the edges of the subunits. By creating substitution rate vectors (Fig. 3B ▶) this picture is clarified greatly. On the whole, the most preserved sides of the helices (indicated by the arrow) face the inside of helical bundles from the same polypeptide chain. As with bacteriorhodopsin, the oligomerizing surfaces show no special evolutionary preservation. Only two protein examples are shown here using the residue color method, as it is simpler to represent the overall trend for the whole database by the vector correlations presented below.
Fig. 2.

The transmembrane residues within bacteriorhodopsin (1c3w) shaded according to estimated substitution rate index Ri (A) and conservation Ci (B). The delineated helices are shown (upper right) for clarity. Within the shading scales, black indicates residues which are the most preserved during evolution and white those which are most variable.
Fig. 3.
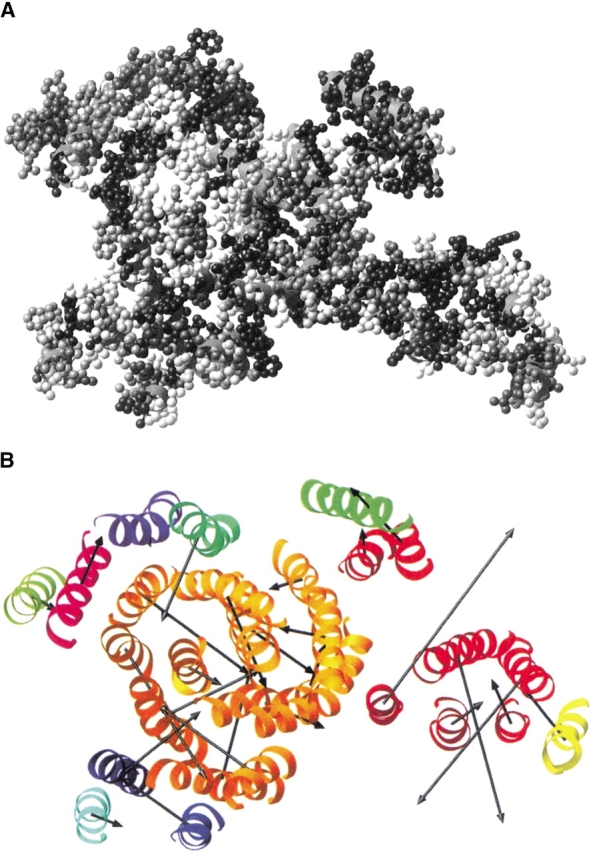
The transmembrane helices of cytochrome c oxidase with residues colored according to estimated substitution rate index Ri (A; as in Fig. 4 ▶) and with superimposed substitution rate vectors (B). For the vector analysis, each subunit of the structure is colored differently.
Substitution rate and lipid exposure
For all the proteins in the structural database, potential correlations of the substitution rate and conservation vectors with the solvent-accessible surface are shown by generating vector dot products, shown in Figures 4 and 5 ▶ ▶. The dot product is the parallel projection of one vector property on another. Hence, the dot product will be large and positive if a vector is coincident with the accessibility vector, large and negative if the vectors are anti-parallel, and near zero if the vectors are perpendicular or if one vector has a small magnitude. In this case, positive dot products indicate that the most evolutionarily variable face of a helix is on the same side as the lipid-accessible face. Overall, in Figures 4 and 5 ▶ ▶ the shape of the dot product distributions are modeled reasonably well by the random distribution. However, there are some obvious differences to the null hypothesis; these differences depend on the use of different accessibility and evolutionary variability measure combinations. All of the histograms show some indication of an evolutionarily preserved core in transmembrane domains; relative to the random distribution, there is an absence of negative dot products and an over-abundance of positive dot products. The bias for positive dot products is more distinct when using estimated substitution rates (Ri) as compared to conservation (Ci). It should be noted that no improvement of this trend is found for Ci if it is expressed as a value relative to the helical average, as is done for Ri. For a given type of residue variability measure (Ri or Ci), using a single polypeptide chain in the accessibility calculation shows a greater bias for positive dot products than using the native oligomeric structure. This is particularly noticeable in the conservation analysis. The combination of single-chain accessibility and relative substitution rate generates the most non-random distribution (Fig. 5B ▶). Indeed, there is only one significant substitution rate vector pointing away from the core of the polytopic transmembrane polypeptides in the data set of helices studied. This single exception is for the helix corresponding to residues 931–950 of the Ca2+ ATPase (1eul). Many of the dot products that are large and negative in the single-chain calculation are near zero when the native oligomeric structures are used. This is consistent with helices at oligomer interfaces having small native accessibility vectors, but large single-chain accessibility vectors. Finally, Figure 6 ▶ shows the correlation between the oligomerizing surface area and substitution rate. Most helices in the data set do not have any oligomerizing surface and thus are not represented. Overall, the dot products fit the null (i.e., random) hypothesis. However, when the substitution rate vectors have their largest magnitude they are opposite to the oligomer interface.
Fig. 4.

The distribution of dot products between the conservation vector and native-chain accessibility vector (A) or single-chain accessibility vector (B). Dot products are positive if vectors are coincident with the accessibility vector, negative if the vectors are anti-parallel, and near zero if the vectors are perpendicular or if one vector has a small magnitude. The absolute values of the dot products (left) and the distribution expected if helices were oriented randomly (solid line ). The difference between the observed dot product data and the random distribution (right). The dotted lines and error bars represent the width of one standard deviation for a random data set with the same number of data points as the structural data. Note that the magnitude of the conservation vectors has been scaled linearly by a factor of −1.6 to give histograms directly comparable with those in Figure 7 ▶; ordinarily, the conservation vector would be in the opposite direction to the substitution rate vector.
Fig. 5.

The distribution of dot products between the substitution rate vector and (A) native-chain accessibility vector or (B) single-chain accessibility vector. The absolute values of the dot products (left) and the distribution expected if helices were oriented randomly (solid line ). The difference between the observed dot product data and the random distribution (right). The dotted lines and error bars represent the width of one standard deviation for a random data set with the same number of data points as the structural data.
Fig. 6.

The distribution of dot products between the substitution rate vector and the hetero-oligomer contact vector. In the left hand chart the dotted line indicates the null hypothesis distribution. The chart (right) shows the difference between the observed dot-product data and the null distribution.
Discussion
Helix–helix interfaces
Earlier observations on the photosynthetic reaction center (Donnelly et al. 1993) indicated that the lipid-exposed regions of transmembrane proteins were less well-conserved during evolution. The distribution of the substitution rates for buried and exposed residues (Fig. 1 ▶) and the accessibility vector correlations (Figs. 4, 5 ▶ ▶) for whole helices confirm this trend and also provide a more significant statistical basis that this is a property of polytopic membrane proteins as a class.
Comparing Figures 4A and 5A ▶ ▶ with Figures 4B and 5B ▶ ▶ illustrates how the less variable residues predominate at the helix–helix interfaces of individual polypeptide chains. The inclusion of multiple polypeptide chains in the accessibility calculation appears to obscure the correlation between estimated substitution rate (ESR) vectors and the core residues of individual polypeptides. Investigation of the oligomerizing interfaces with a vector analysis (Fig. 6 ▶) illustrates that they are not significantly preserved during evolution. Indeed, if significant, there is a bias for the most evolutionarily preserved face of a helix to be on the opposite side to the contact. This observation of helical properties is consistent with the single residue analysis presented in Figure 1 ▶. Here, the distribution of ESR values for oligomer-contacting residues resembles that of lipid-exposed residues most closely. As a class, the core residues (mostly at same chain interfaces) are more highly preserved than the oligomer-interface residues. These data indicate that there is a difference in the selective pressures imposed on same-chain and (mostly hetero-) oligomerizing interfaces. One may infer that within the transmembrane helices of a single polytopic membrane protein, helix–helix interactions are preserved to maintain proper folding. In contrast, for oligomerization the helices need not be so conserved, except for cases in which the subunit interfaces form a bio-functional environment (see below). This indicates that the specificity of these oligomerizing interactions is mediated more by extramembranous elements. It is notable that, within the constraints of hydrophobicity, changing a transmembrane residue at a hetero-oligomerizing helix interface would not be expected to affect the folding of the polypeptide in the initial instance (i.e., the surfaces are separate and lipid-solvated.)
Structure and functionality
The analysis presented here may seem to be at odds with the well-accepted notion that the oligomerizing interfaces of particular homo-oligomeric transmembrane domains must be well conserved. For example the glycine-rich dimerization motif of glycophorin has been shown readily by mutagenesis (Lemmon et al. 1992, 1994). Also, the mutagenesis of residues in ion channel proteins shows that loss-of-function substitutions involve residues within the channel pore or at helix interfaces (i.e., in the core; Arkin et al. 1994; Adams et al. 1995). However, the data set used here is dominated by large hetero-oligomeric protein complexes. Hence, the oligomerization occurs predominantly between helices with different amino acid sequence, rather than between the identical helices of functional homo-oligomers. Reclassification of the few homo-helix interactions in the data set as same-chain interactions gives a very slightly increased bias for a preserved core, but the significance cannot be judged on so few interactions. The distinction between a conserved homo-oligomer interface and the generally undiscerning oligomer interfaces presented here may reflect merely where the functionality of a protein lies. As the primary constraint on the transmembrane amino acid sequence will be fulfilling the biological function of the protein, the placement of residues required to create the biologically active environment will be preserved. This environment can be formed either by an oligomerization (e.g., phospholamban) or, as predominates here, by the association of helices within a polytopic bundle (e.g., bacteriorhodopsin). Hence, the conserved polytopic cores illustrated here may reflect the requirement for a functional scaffold.
Estimated substitution rates
Of particular importance in this study is the use of substitution rates together with sequence similarity to give an indication of evolutionary variability. Given a significant data set, substitution rate analysis is by its nature insensitive to the choice of sequences aligned for evolutionary comparison. Also, as we have shown, a substitution rate analysis can show details of biological interest better than a similarity analysis. In this instance, substitution rates may provide better parameters for modeling transmembrane domain structures. A good example is the picture of bacteriorhodopsin (Fig. 2 ▶) as the picture generated by conservation values can be attributed to maintenance of functional residues, but the more graduated distribution of substitution rate shows a discernable structural influence.
Part of the reason for the difference between conservation and substitution rate may reflect the fact that ESR analysis has a built-in expectation of how many differences will occur. For conservation, it cannot be judged whether a large variation in a sequence element is expected as a result of the particular sequences chosen. Thus, conservation cannot provide as much sensitivity among the more variable residues. It is obvious that, for a given protein, closely related sequences have fewer residue differences than more distantly related ones; indeed this is the expectation of ESR analysis. ESR data will show any significant bias for a residue to change or be preserved relative to this base line. For structural analyses, estimation of substitution rates is like site-directed mutagenesis, in that the tolerance of a site for change is measured, rather than the similarity in the sequences chosen. A similarity analysis can miss important information, as illustrated in Figure 7 ▶.
Fig. 7.

An illustration of sequence elements in two distinct evolutionary situations. A simple similarity analysis cannot distinguish between the higher (left) and lower substitution rate (right).
Also important is that transmembrane helices always carry a significant complement of hydrophobic residues, but this conservation of hydrophobic character is not under investigation as ESR analysis treats all substitutions equally. Thus, conservation data may mask the structurally significant differences in substitution rate.
Conclusions
Polytopic membrane proteins evolve to preserve residues in the cores of a particular polypeptide chain. Residues at oligomer interfaces, except perhaps when there is a direct involvement in biochemical processes, are less well-preserved. Hence, the means of interaction between oligomerizing transmembrane proteins may in general be mediated by extramembranous protein elements. These observations are of particular relevance if molecular modeling of transmembrane proteins is to consider larger, oligomeric complexes. Substitution rates may be used to provide constraints and rationalization for the orientation of helices of a particular polypeptide chain, but there is no statistical foundation indicating that this method can be used to predict the contacts between chains of different sequence.
For a significant number of aligned sequences, ESR analysis provides a rigorous means of generating a picture of substitution rate; it is at least comparable to similarity measurements, but is relatively unbiased by sequence selection and potentially more sensitive than a conservation analysis. Thus, ESR analysis may be applied well for subjects other than hydrophobic transmembrane domains for which an unbiased overview of site-specific evolution is required, especially if the residue characteristics are otherwise conserved.
Materials and methods
Protein structure database
The membrane protein structures used in this analysis represent all of the currently available, non-homologous high resolution structures within the PDB which traverse a lipid bilayer with one or more hydrophobic α-helices and have a sufficient number of sequenced homologues (see below). This analysis is not concerned with the structures of β-barrel, transient, or amphipathic membrane proteins. The structures studied range in size from the two helices of a glycophorin A to the 54 helices of the cytochrome c oxidase. The proteins also have diverse functions. The database used includes the following proteins (PDB identifiers are indicated in parentheses):
Bos taurus Cytochrome c oxidase (1occ) (Tsukihara et al. 1996)
Gallus gallus Cytochrome bc1 complex (1bcc) (Zhang et al. 1998)
Halobacterium halobium Bacteriorhodopsin (1c3w) (Luecke et al. 1999)
Rhodopseudomonas acidophia Light harvesting complex (1kzu) (Fyfe and Cogdell 1996)
Homo sapiens Glycophorin A (1afo) (MacKenzie et al. 1997)
Rhodopseudomonas viridis Photosynthetic reaction center (1prc) (Deisenhofer et al. 1995)
Streptomyces lividans Potassium channel (1bl8) (Doyle et al. 1998)
Escherichia coli Succinate dehydrogenase (1fum) (Iverson et al. 1999)
Oryctolagus cuniculus Ca2+ transporting ATPase (1eul) Toyoshima et al. 2000)
Bos taurus Rhodopsin (1f88) (Palczewski et al. 2000)
These structures represent 80 distinct transmembrane helices containing a total of 1430 residues. The structures of the transmembrane domains of the above proteins were obtained by selecting the bilayer-traversing subset of the PDB structures. This delineation was performed in the same manner as previous analyses (Stevens and Arkin 1999), with automated hydropathy searching and helical secondary structure selection together with a manual check of each structure. To avoid over-representation, in which PDB structures consist of homo-oligomers, only one of the identical subunits was used in further analyses.
Sequence preparation
Sequence selection
For each of the proteins in the high-resolution structural databases, the amino acid sequence was used to find evolutionary variant sequences of the same type of protein. The sequence from the PDB structure was compared for homology with the sequences from the SPTR sequence database, which contains SWISSPROT (Bairoch and Boeckmann 1991) sequences and translated EMBL (Stoesser et al. 1999) sequences, as well as the translated open reading frames (proteomes) of all those organisms which have had their entire genomes sequenced. It should be noted that these databases are not mutually exclusive; repeat sequences were ignored during the analyses. Searches of the SPTR and proteomes databases used the FASTA (Pearson and Lipman 1988) algorithm. Of those homology matches identified for the PDB-derived query sequences, only those with a random match probability <1 in 105 were chosen for further analysis. When large numbers of homologues were identified, only the best 100 with the closest match of their functional description to the query protein were used. For each membrane protein sequence a multiple sequence alignment of the homologues was created using the PILEUP program of the GCG Wisconsin package. Once aligned, hydropathy analysis was performed on each of the sequences to delineate transmembrane regions. This was done using very permissive search parameters, a window of 15 residues and a hydropathy threshold of −22 kCal mole−1 (Stevens and Arkin 2000). Any sequence that did not possess a hydrophobic putative-transmembrane region which overlaps (according to the alignment) each of those predicted from the PDB-derived sequence was removed from consideration. This filtering eliminated significantly truncated homologues and non-membrane variants.
Phylogenetic trees
Using the multiple sequence alignments as input, evolutionary distance matrices were calculated using the PROTDIST programs of the PHYLIP package (Felsenstein 1989); then evolutionary trees were calculated using the FITCH program of the same package. The global alignment used to calculate the evolutionary distance matrix employs the Dayhoff PAM 250 matrix scores (Dayhoff et al. 1983). A more sophisticated alignment scheme was believed unnecessary, as the sequences are already known to share a high degree of homology. Evolutionary tree generation used the Fitch-Margoliash algorithm (Fitch and Margoliash 1967) and considered global rearrangements to give the best possible tree.
Substitution rate
Comparison of residues at a single alignment position is used to estimate the rate of amino acid substitution, given the phylogenetic tree generated by global comparison. In a multiple sequence alignment of different homologues of a protein, for each residue position i the ESR si is calculated as the ratio between the estimated number of substitutions and the total evolutionary distance sampled:
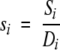 |
1 |
Assuming a given evolutionary tree for the alignment, Si is the minimum number of substitutions required to give rise to the sequence variation of the alignment at position i and Di is the total evolutionary distance spanned at the same position.
The number of substitutions Si required to generate the sequence found in an alignment, according to the phylogenetic tree, is estimated by assuming parsimony. Given that the total number of possible amino acid transitions is much higher than for single nucleotide transitions, the probability of a sequential pair of reciprocating transitions (X → Y → X) is assumed to be negligible. Substitutions were counted in a regressive manner from the outside of the phylogenetic tree (i.e., the actual sequences) towards the central nodes. By working inwards, it is possible to predict the sequence at each node of the tree. If the branches that emanate from a node differ in amino acid (and their assignment is unambiguous), a substitution is scored. A sequence element can only be assigned for each trigonal node once sequence elements of two of its branches are known (see Fig. 8 ▶). The assignment and scoring of substitutions were done according to the scheme presented in Table 1.
Fig. 8.

The scoring of substitutions given a phylogenetic tree of globally aligned protein sequences. The diagram shows the scoring at a single amino acid position. (scoring of a substitution by asterisks).
Table 1.
The scoring scheme used to estimate the occurrence of substitutions at the nodes of a phylogenetic tree
| Node branch 1 | Node branch 2 | Node assignment | Substitution scored? |
| A | A | A | No |
| A | B | A or B | Yes |
| A or B | A | A | No |
| A or B | A or B | A or B | No |
| A or B | A or C | A | No |
| A or B | C | Undefined | Yes |
| A or B | C or D | Undefined | Yes |
| A | Undefined | A | No |
| Undefined | Undefined | Undefined | No |
A, B, C, and D represent different, unspecified amino acid residues estimated to occur at the nodes of a phylogenetic tree.
Total evolutionary distance Di is calculated as the sum of the evolutionary distances di over the paths j of the phylogenetic tree for one position i in the sequence alignment. Note that total path-length varies according to alignment position because of the presence of gap insertions; that is, certain branches of the phylogenetic tree are absent if a sequence has no representative residue at that alignment position (di,j = 0). However, this is rare in transmembrane domain sequences. For a total of J branches,
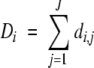 |
2 |
The substitution rate index Ri provides a measure of the deviation of the substitution rate at a single position from the average rate for the whole protein alignment. In this way, all indices (Ri) are relative to a normalized value for the alignment. Hence, different proteins can be compared in a consistent manner. The average substitution rate 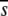 is determined from the total number of substitutions estimated to occur over all the evolutionary distance represented by the phylogenetic tree at all alignment positions.
is determined from the total number of substitutions estimated to occur over all the evolutionary distance represented by the phylogenetic tree at all alignment positions.
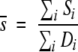 |
3 |
Ri is calculated by the expression:
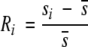 |
4 |
Hence, Ri has a lower limit of −1 when there are no substitutions. In the sequence data used here there were no values of Ri >4. Also, for transmembrane residues Ri was very rarely >3. In subsequent analyses, for example to generate helical vectors (see below), Ri may be used as is or expressed relative to the average Ri for a given helix.
Minimum number of nodes
It should be noted that Ri can be generated only for alignments with sufficient protein sequences. With a small number of sequences, the number of nodes of the evolutionary tree is also small. Hence, the substitution events will be sampled poorly. Over large evolutionary distances, intermediate substitutions may be missed and, for small distances, substitutions may have undue significance. In other words, the substitution rate will be anomalously high if only a single substitution event has occurred and anomalously low if no substitution has occurred. The average probability of finding a substitution event over all the sequence alignments of the proteins studied here is ∼16%, so as a rough guide we have estimated the minimum number of nodes for a representative phylogenetic tree to be ∼25. At this level, the difference of a single substitution will change the substitution rate by ±4%. Thus, the mean ±1 substitution (16% ± 4%) is at least 2 substitutions away from 0 + 1 substitution. For 10 nodes, 16 ± 10% is indistinguishable from 0 + 10%.
Similarity analysis
A more traditional conservation analysis of the sequence alignments was done using the PLOTSIMILARITY program of the GCG Wisconsin package considering a window of a single residue. As with the ESR analysis, this generates a value for the variability of the amino acids at each position in the alignment. The conservation value at each position Ci is used in subsequent analysis in the same manner as Ri.
Visualization
For positions in the alignment corresponding to the delineated helices (as used in the hydropathy check), the ESR indices and conservation values were tabulated to find any structurally related patterns of evolutionary variation. The residues in a pictorial representation of the three-dimensional transmembrane domain structure were colored according to ESR index and conservation, as shown in Figures 2 and 3 ▶ ▶. The color used for each residue was a shade between variable (red) and conserved (blue), in which the proportion of red and blue color components increase and decrease, respectively, in a linear manner according to Ri or Ci.
Also, for each of the transmembrane helices, accessibility was compared to the ESR index and conservation by creating helical property vectors. A helical property vector shows the overall direction and relative magnitude of a given residue property in a helix. Accordingly, ESR index, conservation, and accessibility vectors were calculated to indicate which face of a helix has the lowest substitution rate, is most conserved, and most solvent-accessible, respectively. Each helix vector is the sum of its residue vectors resolved in a plane orthogonal to the axis of the helix. The direction of each residue vector is parallel to the vector from the α-carbon position to the geometric mean of the side chain and the magnitude of the vector is simply proportional to the property under investigation (e.g., Ri). Two types of accessibility value were calculated using the output of the program MSRoll (Gerstein et al. 1995). The first is the native accessibility; the solvent-exposed surface area of residues in the complete PDB structure. This includes cofactor atoms and ions but excludes small molecules required for crystallization (e.g., lipids and sulfate anions). The second is the single chain accessibility, which is the solvent exposed surface area of residues of a single polypeptide chain in isolation (see Fig. 9 ▶). Lastly, oligomer-contacting surfaces were calculated using these accessibility values; the single chain accessibility is greater than the native accessibility because of the exposure of oligomerizing surfaces to the solvent accessibility calculation. Hence, the oligomerizing surface area is calculated as the difference between single chain accessibility and the native accessibility. As with the other types of surface, the oligomerizing areas can be used to generate helix vectors, in this instance to indicate which face of the transmembrane helices touch other chains.
Fig. 9.
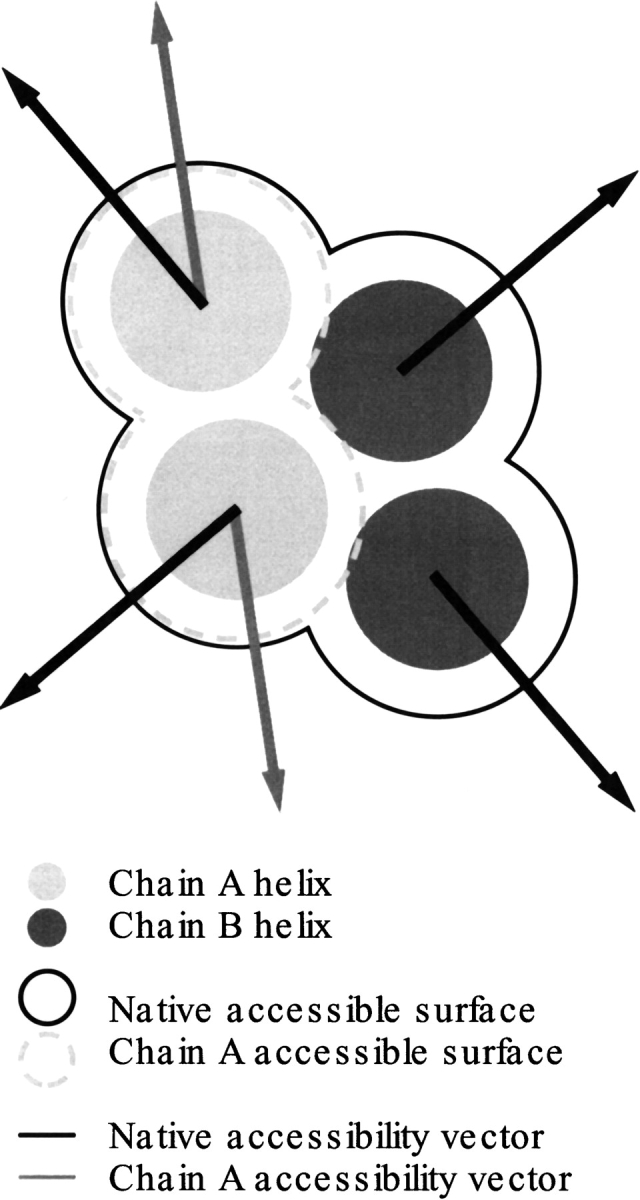
The two types of accessibility calculation used to generate accessibility vectors.
Dot products
Dot products were calculated to compare the relative magnitude and direction of the ESR and conservation vectors with the accessibility vectors. This calculation is done to generate a measure of coincidence of the vector properties. These data were then used to generate histograms, showing the distribution of dot products for all helices in the database. The dot products for the helix vectors were compared with the null hypothesis distribution, which considers vectors to be oriented randomly. This null distribution is generated from the dot product between two real helix property vectors, in which one is rotated in increments through 360°. To generate a smooth and representative distribution, dot products for each pair were calculated for 10,000 orientations. Given the relatively limited number of helices in the data set, the dot products have a degree of sampling noise. The potential influence of the sample size on the histograms was determined by calculating the standard deviation of randomly oriented helices. Accordingly, a large number of random data sets were generated that are equal in size to the real helix set. These dot products were partitioned into the same histogram bins as the structural data to estimate the width of the random distribution at each bin. The standard deviations thus calculated were used to illustrate the confidence bounds of the histogram data.
Acknowledgments
This work was supported by grants from the Wellcome trust and the Biotechnology and Biological Sciences Research Council to I.T.A.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.10501.
References
- Adams, P.D., Arkin, I.T., Engelman, D.M. and Brunger, A.T. 1995. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat. Struct. Biol. 2 154–162. [DOI] [PubMed] [Google Scholar]
- Adams, P.D., Engelman, D.M. and Brunger, A.T. 1996. Improved prediction for the structure of the dimeric transmembrane domain of glycophorin A obtained through global searching . Proteins Struct. Func. Gen. 26 257–261. [DOI] [PubMed] [Google Scholar]
- Arkin, I.T., Adams, P.D., MacKenzie, K.R., Lemmon, M.A., Brünger, A.T. and Engelman, D.M. 1994. Structural organization of the pentameric transmembrane α-helices of phospholamban, a cardiac ion channel. EMBO J. 13 4757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin, I.T., MacKenzie, K.R. and Brunger, A.T. 1997. Site directed dichroism as a method for obtaining rotational and orientational constraints for oriented polymers. J. Amer. Chem. Soc. 119 8973–8980. [Google Scholar]
- Bairoch, A. and Boeckmann, B. 1991. The SWISS-PROT protein-sequence data-bank. Nucleic Acids Res. 19(SS):2247–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, F.C., Koetzle, T.F., Wiliams, G.JB., Meyer (Jr), E.F., Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T. and Tasumi, M. 1977. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 112 535–542. [DOI] [PubMed] [Google Scholar]
- Dayhoff, M.O., Barker, W.C. and Hunt, L.T. 1983. Establishing Homologies in Protein Sequences. Methods Enzymol. 91 524–545. [DOI] [PubMed] [Google Scholar]
- Deisenhofer, J., Epp, O., Sinning, I. and Michel, H. 1995. Crystallographic refinement at 2.3 Å resolution and refined model of the photosynthetic reaction centre from Rhodopseudomonas viridis. J. Mol. Biol. 246 429–457. [DOI] [PubMed] [Google Scholar]
- Donnelly, D., Overington, J.P., Ruffle, S.V., Nugent, J.H.A. and Blundell, T.L. 1993. Modeling alpha-helical transmembrane domains—The calculation and use of substitution tables for lipid-facing residues. Protein Sci. 2 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, D.A., Cabral, J.M., Pfuetzner, R.A., Kuo, A., Gulbis, J.M., Cohen, S.L., Chait, B.T. and MacKinnon, R. 1998. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 280 69–77. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. 1989. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 5 164–166. [Google Scholar]
- Fitch, W.M. and Margoliash, E. 1967. Construction of phylogenetic trees. Science 155 279–284. [DOI] [PubMed] [Google Scholar]
- Fyfe, P.K. and Cogdell, R.J. 1996. Purple bacterial antenna complexes. Curr. Opin. Struct. Biol. 6 467–472. [DOI] [PubMed] [Google Scholar]
- Gerstein, M., Tsai, J., and Levitt, M. 1995. The volume of atoms on the protein surface: Calculated from simulation, using voronoi polyhedra. J. Mol. Biol. 249 955–966. [DOI] [PubMed] [Google Scholar]
- Hubbard, T.J.P. and Blundell, T.L. 1987. Comparison of solvent-inaccessible cores of homologous proteins: Definitions useful for protein modeling. Protein Eng. 1 159–171. [DOI] [PubMed] [Google Scholar]
- Iverson, T.M., Luna-Chavez, C., Cecchini, G. and Rees, D.C. 1999. Structure of the E. coli fumarate reductase respiratory complex. Science 284 1961–1966. [DOI] [PubMed] [Google Scholar]
- Lemmon, M.A., Flanagan, J.M., Treutlein, H.R., Zhang, J. and Engelman, D.M. 1992. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 31 12719–12725. [DOI] [PubMed] [Google Scholar]
- Lemmon, M.A., Treutlein, H.R., Adams, P.D., Brunger, A.T. and Engelman, D.M. 1994. A dimerization motif for transmembrane alpha-helices. Nat. Struct. Biol. 1 157–163. [DOI] [PubMed] [Google Scholar]
- Luecke, H., Schobert, B., Richter, H-T., Cartailler, J-P. and Lanyi, J.K. 1999. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291 899–911. [DOI] [PubMed] [Google Scholar]
- MacKenzie, K.R., Prestegard, J.H. and Engelman, D.M. 1997. A transmembrane helix dimer: structure and implications. Science 276 131–133. [DOI] [PubMed] [Google Scholar]
- Palczewski, K., Kumasaka, T., Hori, T., Behnke, C.A., Motoshima, H., Fox, B.A., le Trong, I., Teller, D.C., Okada, T., Stenkamp, R.E., [DOI] [PubMed]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popot, J.L. and Engelman, D.M. 1990. Membrane protein folding and oligomerization: the two-stage model. Biochemistry 29 4031–4037. [DOI] [PubMed] [Google Scholar]
- Sakai, H. and Tsukihara, T. 1998. Structures of membrane proteins determined at atomic resolution. J. Biochem. 124 1051–1059. [DOI] [PubMed] [Google Scholar]
- Stevens, T.J. and Arkin, I.T. 1999. Are membrane proteins "inside-out" proteins? Proteins Struct. Func. Gen. 36 135–143. [DOI] [PubMed] [Google Scholar]
- Stevens, T.J. and Arkin, I.T. 2000. Do more complex organisms have a greater proportion of membrane proteins? Proteins 39 417–420. [DOI] [PubMed] [Google Scholar]
- Stoesser, G., Tuli, M.A., Lopez, R. and Sterk, P. 1999. The EMBL Nucleotide Sequence Database. Nucleic Acids Res. 27 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima, C., Nakasako, M., Nomura, H. and Ogawa, H. 2000. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Angstrom resolution. Nature 405 647–655. [DOI] [PubMed] [Google Scholar]
- Treutlein, H.R., Lemmon, M.A., Engelman, D.M. and Brunger, A.T. 1992. The glycophorin A transmembrane domain dimer: Sequence-specific propensity for a right-handed supercoil of helices. Biochemistry 31 12726–12732. [DOI] [PubMed] [Google Scholar]
- Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R. and Yoshikawa, S. 1996. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 272 1136–1144. [DOI] [PubMed] [Google Scholar]
- Yamamoto, M. and Miyano, M. 2000. Crystal structure of rhodopsin: A protein-coupled receptor. Science 289 739–745. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Huang, L., Shulmeister, V.M., Chi, Y.I., Kim, K.K., Hung, L.W., Crofts, A.R., Berry, E.A. and Kim, S.H. 1998. Electron transfer by domain movement in cytochrome bc1. Nature 392 677–684. [DOI] [PubMed] [Google Scholar]