

Abstract
Serpins inhibit cognate serine proteases involved in a number of important processes including blood coagulation and inflammation. Consequently, loss of serpin function or stability results in a number of disease states. Many of the naturally occurring mutations leading to disease are located within strand 1 of the C β-sheet of the serpin. To ascertain the structural and functional importance of each residue in this strand, which constitutes the so-called distal hinge of the reactive center loop of the serpin, an alanine scanning study was carried out on recombinant α1-antitrypsin Pittsburgh mutant (P1 = Arg). Mutation of the P10′ position had no effect on its inhibitory properties towards thrombin. Mutations to residues P7′ and P9′ caused these serpins to have an increased tendency to act as substrates rather than inhibitors, while mutations at P6′ and P8′ positions caused the serpin to behave almost entirely as a substrate. Mutations at the P6′ and P8′ residues of the C β-sheet, which are buried in the hydrophobic core in the native structure, caused the serpin to become highly unstable and polymerize much more readily. Thus, P6′ and P8′ mutants of α1-antitrypsin had melting temperatures 14 degrees lower than wild-type α1-antitrypsin. These results indicate the importance of maintaining the anchoring of the distal hinge to both the inhibitory mechanism and stability of serpins, the inhibitory mechanism being particularly sensitive to any perturbations in this region. The results of this study allow more informed analysis of the effects of mutations found at these positions in disease-associated serpin variants.
Keywords: Serpins, protease inhibitor, inhibition, β-sheet, stability
Proteins of the serine protease inhibitor (serpin) superfamily have a common architecture made up of three conserved β-sheets and nine α-helices (Gettins et al. 1996; Whisstock et al. 1998). Most members of the family play the signature role of serine protease inhibition, and as such, control a number of important biological processes, including blood coagulation and inflammation (Potempa et al. 1994; Gils and Declerck 1998). An increasing number of serpins discovered are noninhibitory, and have diverse functions that are often aided by their structure. Mutations in human members of the serpin superfamily frequently contribute to diseases such as thrombosis, angioedema, and emphysema, because loss of function of these regulatory molecules or a deficiency state leads to pathological excesses of activity of their cognate proteases and thereby disease (Stein and Carrell 1995; Carrell and Gooptu 1998).
The inhibitory function of serpins has now been shown to consist of an initial docking of the protease with an exposed reactive center loop (RCL) of the serpin (Stone and Le Bonniec 1997), followed by cleavage of the RCL at the P1–P1′ bond by the protease (as defined by Schechter and Berger [1967], a protease cleaves at the P1–P1′ peptide bond in a substrate; residues N-terminal to the cleavage point are labeled P1, P2, P3, etc., counting outwards from the cleavage point, while residues in the substrate C-terminal to the cleavage point are labeled P1′, P2′, P3′, etc.). In a rapid sequence of events, cleavage of the RCL is arrested at the acyl intermediate stage (Lawrence et al. 1995; Egelund et al. 1998), forming a covalent complex with the protease. This is apparently concomitant with insertion of the RCL into the A β-sheet of the serpin (Shore et al. 1995; Stratikos and Gettins 1997), causing distortion of the proteinase architecture (Stavridi et al. 1996; Kaslik et al. 1997; Huntington et al. 2000; Egelund et al. 2001; Tew and Bottomley 2001). A stable complex of the protease covalently linked to the serpin is thus formed in which the enzyme is found 70 Å away from its initial point of attack at the bottom of the A β-sheet (Huntington et al. 2000). As may be evident, the inhibitory mechanism of the serpins is fraught with "risk" of nonproductive cleavage of the serpin by the protease, resulting in the serpin becoming a proteinase substrate. Thus, the reaction scheme for serpins consists of both inhibitory and substrate pathways, with an inhibitory serpin usually only making use of the former (Gettins et al. 1996).
The RCL of serpins consists of two conserved hinges, identified as the proximal (P10–P15) and distal hinges (s1C of the C-sheet, P6′–P10′), with a variable amino acid sequence between the hinges optimally suited to interaction with cognate proteinases (Stein and Carrell 1995) (Fig. 1 ▶). As the name implies, both hinges are involved in serpin conformational mobility. The proximal hinge must undergo conformational change for the RCL to insert into the A β-sheet during the S-to-R transition. The distal hinge undergoes conformational change during the transition to the latent form (Mottonen et al. 1992), and perhaps as a prerequisite for serpin polymerization (Chang et al. 1997). Mutation of the conserved (usually small aliphatic) amino acid residues of the proximal hinge to more bulky amino acid residues usually results in substrate-like behavior by the serpin, proposed to be due to the RCL not being able to insert into the A β-sheet efficiently enough to inhibit the serpin (Hopkins and Stone 1995). Mutations at the distal hinge also cause perturbation of the serpin inhibitory mechanism, presumably by affecting the conformation of this equally vital hinge of the RCL, and thus interfering with the mechanism (summarized in Stein and Carrell 1995). Mutations within s1C also cause serpins to have an increased propensity to polymerize, as previously seen with C1-inhibitor (Eldering et al. 1995).
Fig. 1.
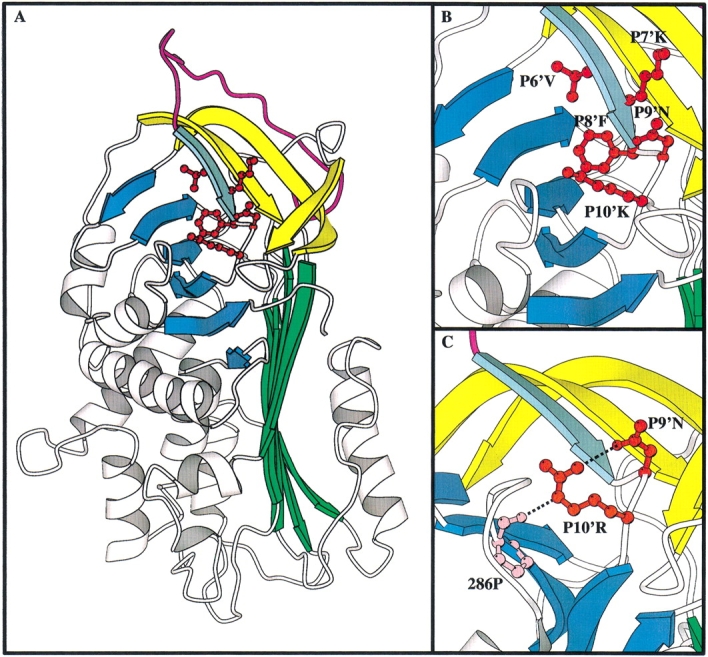
Structural diagram of the C β-sheet mutants of α1-antitrypsin in comparison to the equivalent residues in antithrombin. (A) the structure of α1-antitrypsin (pdb identifier 1QLP; Elliot et al. 1996 1998) with the C β-sheet mutations highlighted. The A β-sheet is in green, the B β-sheet in blue, the RCL in magenta, and strands s2C–s4C of the C β-sheet in yellow. Strand s1C is in aquamarine. P6′–P10′ are shown in red ball and stick. (B) Closeup of the C β-sheet region with the side chains of P6′–P10′ labeled. (C) Closeup on the C β-sheet region in antithrombin (pdb identifier 2ANT; Schreuder et al. 1994; Skinner et al. 1997) showing the hydrogen bond network between P10′R, P9′N, and 286P.
Serpin polymerization is hypothesized to result from inappropriate insertion of the RCL of one serpin molecule into the A or C β-sheet of another, eventually resulting in the formation of long polymers of the molecule. Recent crystallographic structures of cleaved α1-antitrypsin (α1-AT, also known as α1-proteinase inhibitor) have provided some corroboration for this hypothesis for the mechanism of serpin polymerization (Huntington et al. 1999; Dunstone et al. 2000). Polymerization is the primary outcome of heat-induced denaturation of serpins, and can also be potentiated by mutations in serpins, such that they become unstable to temperatures even in the physiological range. The s1C region has been shown to participate in the conversion of serpins to the latent state, in which the RCL is inserted into the A β-sheet without cleavage, as occurs normally for PAI-1 (Mottonen et al. 1992; Tucker et al. 1995) and for some unstable natural mutants of antithrombin (Beauchamp et al. 1998). Thus, anchoring of the C-sheet is critical to prevent the transition to latency (Mottonen et al. 1992) and, in addition, work by Chang et al. (1997) showed that mobility of the C-sheet is required for efficient serpin polymerization. Anchoring of the C-sheet has also been shown to be important to prevent polymerization or latency, and it is thought that "peeling off" of s1C is a prerequisite for both latency and polymerization (Chang et al. 1997).
Several natural variants of serpins with mutations in the s1C of the C-sheet, associated with disease conditions such as thrombosis and angioedema, have been described (Bock et al. 1988; Nakagawa et al. 1991; Lane et al. 1992; Verpy et al. 1995). These molecules apparently have reduced inhibitory activity and an increased tendency to polymerize, causing deficiency states. It is evident, therefore, that this region of the serpin is important for maintenance of stability and inhibitory function, yet no systematic study of the influence of each position on this strand on the inhibition mechanism or stability of the serpin has been carried out to date. Here, we report the results of an alanine-scanning study of the P6′–P10′ positions on serpin stability and function.
Results
Expression and purification of mutant proteins
Alanine scanning mutagenesis of the s1C was performed on the Pittsburgh mutant of α1-AT (P1=Arg), giving rise to five variants. These variants of α1-AT(P1=Arg) are termed P6′Ala, P7′Ala, P8′Ala, P9′Ala, and P10′Ala. The use of the variant of α1-AT containing a P1=Arg allowed us to use thrombin as a cognate protease as this mutant serpin inhibits thrombin very efficiently due to the point mutation at the reactive center. All of the mutants were expressed and purified from E. coli as previously described (Bottomley and Stone 1998). The yields of the wild-type protein and most variants was 20 mg/L of purified protein, while the yields of the P6′Ala and P8′Ala mutants were 2 mg/L. The P6′Ala and P8′Ala mutants could not be expressed at the same level as the other proteins, despite repeated attempts.
Analysis of complex formation by SDS-PAGE
The ability of the α1-AT variants to form SDS-stable complexes with thrombin was examined by incubating 4 μM serpin with 2 μM thrombin for 30 min at room temperature. The resulting mixture was then analyzed by SDS-PAGE under reducing conditions (Fig. 2 ▶). A band with an apparent molecular mass equal to that of thrombin plus variant α1-AT was detected for the variants P7′Ala, P9′Ala and P10′Ala. In addition, a band corresponding to cleaved α1-AT was evident for the P7′ and P9′Ala mutants. The P6′Ala and P8′Ala variants acted as thrombin substrates, and migrated with an apparent molecular mass corresponding to that of α1-AT cleaved within the reactive-site loop after interaction with thrombin.
Fig. 2.

SDS-PAGE analysis of complex formation with thrombin by the S1C mutants of α1-antitrypsin. α1-AT was mixed with thrombin in a 2:1 molar ratio and allowed to incubate for 30 min at 25°C, following which the complexes were analyzed by 10% SDS-PAGE. The mutants analyzed are indicated, with P6′–P9′ mutants and wild type analyzed in (A), while the P10′ mutant is shown in (B). Incubation with thrombin (+) or not (−) is indicated . The positions of complexed, intact, and cleaved α1-AT, as well as thrombin alone, are indicated. The molecular weight markers are shown in lanes marked "M", and their masses are indicated.
Inhibitory properties of the α1-AT variants
Each of the variants of α1-AT was titrated against thrombin, yielding values for the stoichiometry of inhibition (SI) (Table 1). The SI values for the s1C variants against thrombin varied from equivalent to the wild type (1.1) to over 200. The P6′Ala and P8′Ala variants had very high SI values of 150 and 200, respectively. These results are in agreement with those obtained in the SDS-PAGE analysis of complex formation experiments, which indicated that these mutants failed to form SDS-stable complexes, and most of the inhibitor acted as a substrate, yielding bands equivalent to cleaved α1-AT (Fig. 2 ▶). By comparison, the P10′Ala mutant had an SI of 1 and the P7′Ala mutant had an SI of 3.5. The P9′Ala mutant had a relatively high SI of 15; again, these results agree with the data obtained in the SDS-PAGE experiments where the serpin–enzyme complex and cleaved α1-AT bands were observed in ratios consistent with the inhibitory behavior of the mutants. The association rate constants (kass) for the inhibition of thrombin by the variants were determined by progress curve kinetics (Table 1). The value for P10′Ala was similar to that of α1-AT(P1=Arg) inhibition of thrombin, while the kass values for P7′Ala and P9′Ala were 4.6- and 7.7-fold higher than the wild type, respectively, once the SI values for these serpins had been taken into account (Hood et al. 1994). The kass values could not be determined for the even-numbered variants due to their substrate nature.
Table 1.
Kinetic and stability data for C β-sheet mutants of α1-AT compared to wild type
| Protein | SI | kass (×104) M.s−1 | kass (×104) M.s−1 × SI | Tm (°C) |
| P1-Arg-α1-AT | 1.1 | 10 | 10 | 58 |
| P6′ Ala | 177 | substrate | — | 44 |
| P7′ Ala | 3.5 | 13 | 46 | 58 |
| P8′ Ala | 62 | substrate | — | 44 |
| P9′ Ala | 15 | 5.1 | 77 | 58 |
| P10′ Ala | 1.1 | 10 | 10 | 58 |
Stability studies
Previously, mutations in the distal hinge have been shown to cause polymerization of the serpin. This was proposed to be due to a destabilizing of the first strand of the C-sheet, which is required to move for polymerization to proceed. To examine the changes in the stability of our variants, we determined their melting points (Tm) using circular dichroism spectroscopy (Fig. 3 ▶, Table 1). Both P6′Ala and P8′Ala had drastically lowered melting temperatures, while all other mutants had normal melting temperatures relative to the control molecule. Studies using PAGE analysis of the mutants confirmed that high molecular weight polymers were being formed by the P6′ and P8′ mutants (results not shown).
Fig. 3.

Thermal melts of two forms of α1-AT indicating relative molecular stability. The P6′Ala (light gray line) and wild-type α1-AT forms were analyzed by circular dichroism spectroscopy for their stability to thermal denaturation. Thermal unfolding was performed using a heating rate of 60°C/h, and the changes in secondary structure with temperature were measured by monitoring the CD signal at 230 nm.
Discussion
The distal hinge of the RCL of serpins, particularly the residues on strand 1 of the C β-sheet (P6′–P10′), has been shown in a number of separate studies to affect both the inhibitory properties of serpins and their stability. Studies of natural variants with mutations in this region show that the mutant molecules usually have decreased inhibitory capabilities, and also decreased stability (summarized in Stein and Carrell 1995). Thus, mutants of antithrombin at P6′, P8′, P9′, and P10′ have been described (Nakagawa et al. 1991; Lane et al. 1992; Bock et al. 1988), all of which have decreased inhibitory activity and stability (leading to deficiency in antithrombin and thrombotic complications). A mutant of C1 inhibitor at P8′ has also been described that leads to a molecule with similarly decreased inhibitory activity and stability (Verpy et al. 1995). This leads once again to a deficiency state, which in turn, causes angioedema. These natural variants all indicate the importance of this region to the stability and activity of the serpins, but do not constitute a systematic study of the contribution of each residue to the individual properties of stability and inhibitory capability. In addition, the nature of the mutation will have a large influence on the molecular outcome; hence, the need for a systematic study in which a standardized mutation is made at each position, such as that carried out with the alanine scan here.
Studies with recombinant α1-antitrypsin have elucidated an overall contribution of s1C to serpin inhibition and stability. A recombinant mutant of α1-antitrypsin with a disulfide bond engineered into the C β-sheet, which effectively tethered the s1C strand, showed that mobility in the C-sheet was not an essential requirement for the mechanism of serpin inhibition (Hopkins et al. 1997). This is logical in the light of the finding that serpin inhibition results in cleavage of the RCL so that the distal hinge is separated from the rest of the RCL, which then inserts into the A-sheet of the serpin (Huntington et al. 2000). Mobility of s1C was found to be important in the process of serpin polymerization, however, which underlies heat-induced serpin denaturation and loss of function (Chang et al. 1997). Thus, it has been postulated that s1C needs to "peel off" from this sheet during polymerization. Destabilization of the s1C packing in the C-sheet might, therefore, be expected to lead to an increased propensity of the serpin to polymerize, resulting in a lower melting temperature.
It may be seen that mutation of the P7′, P9′, and P10′ residues had very little effect on the stability of the serpins towards heat-induced polymerization, because their melting temperatures were identical to the wild type (Table 1). All the above proteins have mutations to residues that point away from the body of the molecule; thus, it appears that mutations at these positions have no effect on the "anchoring" of s1C to the C-sheet and, therefore, do not destabilize the molecule as a whole. In contrast, mutants at the P6′ and P8′ positions had drastically reduced melting temperatures relative to the wild type, indicating a strong propensity to polymerize. This would indicate that destabilization at these positions causes increased mobility of s1C, which allows this strand to peel off more easily from the C-sheet and thus induce polymerization at much lower temperatures. Both P6′ and P8′ are hydrophobic residues buried in the core of the molecule, and thus most likely constitute primary anchorage points for s1C, limiting the mobility of this strand and thus contributing to the stability of the serpin.
The results of our study indicate that mutating several positions in s1C to alanine had some effect on the serpin inhibition mechanism. Large effects were seen in the P6′ and P8′ positions, a relatively moderate effect was seen at P9′, and a smaller effect was seen at P7′ (Table 1). Mutation of the P10′ residue had no apparent effect on the inhibitory capability of the serpin. The P7′ and P9′ Ala mutants were affected in terms of both their partitioning between substrate and inhibitory paths and the rate of association between the serpin and enzyme (kass). The kass values for P7′Ala and P9′Ala were somewhat increased (once SI had been taken into account [Hood et al. 1994]), indicating that presentation of the RCL in the initial complex between serpin and enzyme was more favorable for both mutants. Interestingly, the increase in the association constant appeared to be proportional to the increase in SI, indicating that, although presentation of the RCL was more favorable in the mutants, there was a concomitant perturbation of the partitioning between substrate and inhibition pathways due to the mutations. It is not easy to explain these results in structural terms, because neither the P7′ Lys residue nor the P9′Asn residue appear to be participating in any bonding with other residues. These outward-facing residues must, therefore, be playing some more subtle, yet important, role in the formation of the initial complex with the protease and in the progression to the trapped acyl intermediate. The effect on SI was seen very strongly for the P6′Ala and P8′Ala mutants. It appears that the substantial changes induced by changing residues that form part of the hydrophobic core in these mutants causes a major disruption to the serpin inhibitory mechanism, resulting in the mutant molecules behaving almost completely as substrates rather than inhibitors. Cumulatively, the effects of the mutations in s1C on the inhibition mechanism indicate that maintenance of the stability of this strand of the C-sheet is vital to maintain partitioning between substrate and inhibition pathways in the serpin mechanism. In the light of the recent determination that the RCL in a serpin is cleaved during inhibition and the protease translocates to the bottom of the A-sheet (Huntington et al. 2000), it is perhaps not simple to see why s1C mutations should affect the inhibitory mechanism. Our simplest interpretation of the results is that, despite the distal hinge being left behind at the top of the molecule after RCL cleavage, any perturbation of the way in which the RCL is held during the rapid interaction with the protease results in an increased cleavage of the serpin, instead of inhibition occurring. This, in turn, substantiates the hypothesis put forward by Stein and Carrell (1995), that absolute maintenance of the correct positioning of the hinges of the RCL must be maintained to effect the inhibitory mechanism of serpins.
In light of the results in the above study, it is of interest to re-examine results with natural variants of serpins at these positions that are associated with disease. Antithrombin mutants at P6′ and P8′ and a C1 inhibitor mutant at P8′ all cause deficiency and loss of inhibitory activity (Bock et al. 1988; Lane et al. 1992; Verpy et al. 1995). This is in line with our results indicating that these positions are indeed critical to the maintenance of packing of s1C into the C-sheet, and that serpins with mutations at these positions behave as substrates and have drastically lowered stability. There are no known disease states associated with natural variants at the P7′ position (Stein and Carrell 1995), again concordant with our results, indicating that this position only has minor effects on the inhibitory mechanism of serpins and no effect on the stability. A mutant of C1 inhibitor at the P9′ position had no molecular effects (possibly due to the reasonably conservative nature of the Gln to Glu substitution) (Verpy et al. 1995), but in antithrombin La Rochelle, an Asn-to-Lys mutation at P9′ caused deficiency and decreased inhibitory activity (Lane et al. 1992). The P9′Ala mutant studied here did indeed have altered inhibitory capability, primarily due to a large increase in its SI value, but had normal stability. Similarly, while antithrombin Kyoto P10′(Arg to Met) had decreased inhibitory activity and led to a deficiency state (Nakagawa et al. 1991), the P10′Ala mutant was completely normal. The discordance between our results with α1-antitrypsin and those found in natural variants of antithrombin indicate that the organization of residues in the s1C of antithrombin might differ at this point compared to α1-antitrypsin, thus yielding the apparent contradiction with the results obtained here. Close examination of the crystal structures of antithrombin (Skinner et al. 1997) and α1-antitrypsin (Elliott et al. 1996, 1998) indicates that there are indeed differences at the P9′ and P10′ positions, primarily the existence of a hydrogen bonding network in antithrombin, which is absent in α1-antitrypsin (Fig. 1 ▶). At first glance, the proteins appear to be highly similar at these positions, with an Asn residue at P9′ for both antithrombin and α1-antitrypsin and a Lys residue at P10′ in α1-antitrypsin compared to an Arg residue at the equivalent position in antithrombin (Fig. 1B,C). However, the arginine residue at P10′ in antithrombin is making hydrogen bonds that the corresponding lysine in α1-antitrypsin is not. Thus, the Nɛ of the Arg residue in antithrombin hydrogen bonds to the backbone oxygen of the Pro286 (Pro255 in α1-antitrypsin) residue on strand 3 of the B β-sheet and the guanidino N hydrogen bonds to the side chain of the P9′Asn residue (Fig. 1C ▶). The Lys residue found in α1-antitrypsin does not possess the guanidino N or the Nɛ groups, and the X-ray crystal structure (Elliot et al. 1996 1998) reveals no hydrogen bonds with Pro286 or the side chain of the P9′Asn residue (Fig. 1B ▶). Thus, the differences noted at P9′ and P10′ between natural variants of antithrombin and the mutants of α1-antitrypsin produced here appear to be explicable in terms of the absence of a hydrogen bonding pattern in α1-antitrypsin, which is apparently quite vital in antithrombin.
The systematic substitution of alanine residues at every position of the s1C strand in the C-sheet of α1-antitrypsin has allowed us to clearly delineate the contribution of each position to serpin stability and function. Predictably, changes at P6′ and P8′ positions had large effects on the molecules, most likely due to large disturbances to the packing of the side chains facing into the body of the molecule at these points. Mutations of residues with side chains facing away from the body of the molecule (P7′, P9′, and P10′) did not affect stability, although P7′ and P9′ mutations did have some effect on inhibitory activity. This indicates that anchoring of s1C is vital to the maintenance of serpin inhibitory function, in particular the partitioning of the serpin between substrate and inhibitory pathways. Cumulatively, the results allow more reasoned interpretation of the effects of s1C mutations, and provide a template for interpretation of results obtained with natural variants associated with disease.
Materials and methods
Materials
Restriction enzymes were obtained from New England Biolabs, and oligonucleotides were purchased from Geneworks. The chromogenic substrate D-Val-Leu-Arg-p-nitroanilide (S-2266) was obtained from Chromogenix. Thrombin was purified from time-expired human plasma as described previously (Stone and Hofsteenge 1986). All other chemicals and materials were obtained from Sigma.
Mutagenesis and expression
The s1C variants were produced using α1-AT(P1=Arg) as the template. PCR methodology was used to generate the alanine mutants. The 5′ primer was designed to encompass the AvaI restriction site, which lies at a position corresponding to codons for residues P4' and P5’ in addition to the site of mutagenesis. The 3′ primer corresponded to the end of the gene followed by a BamHI site. PCR fragments were digested with AvaI and BamHI, and ligated into similarly cut vector coding for α1-AT(P1=Arg). The presence of the desired mutation and the absence of others were confirmed by sequencing of the gene. The α1-AT variants were expressed and purified as previously described (Bottomley and Stone 1998). The purified protein was snap frozen in liquid N2 and stored at −70°C. The concentrations of the variants were estimated from their absorbance at 280 nm using an extinction coefficient (ɛ1%) of 5.8 cm−1.
Analysis of complex formation
The ability of the α1-AT variants to form SDS-stable complexes with thrombin was assessed by incubating the variants (4 μM) with thrombin (2 μM) in 50 mM Tris–HCl buffer, pH 7.8 containing 150 mM NaCl for 30 min at 25°C. This incubation mixture was then analyzed by 10% SDS-PAGE. The following molecular weight markers were used: myosin, 200 kD; β-galactosidase, 116.3 kD; phosphorylase b, 97.4 kD; bovine serum albumin, 66.3 kD; glutamate dehydrogenase, 55.4 kD; lactate dehydrogenase, 36.5 kD; carbonic anhydrase, 31 kD.
Determination of stoichiometry of inhibition and association rate constant
The stoichiometry of inhibition and association rate constant for the variants against thrombin were determined as previously described (Stone and Hermans 1995). The concentration of thrombin was determined by active site titration with p-nitrophenyl p’-guanidinobenzoate (Chase and Shaw 1969).
Circular dichroism
Circular dichroism (CD) spectra were measured on a Jasco 820s spectropolarimeter at 25°C. Far-UV spectra from 190– 250 nm were collected with 5 sec/point signal averaging. Thermal unfolding was performed using a heating rate of 60°C/h, and the changes in secondary structure with temperature were measured by monitoring the CD signal at 230 nm and a protein concentration of 0.05 mg/mL in 50 mM Tris, 50 mM NaCl, pH 7.8. The temperature within the cuvette was maintained by a computer-controlled water bath connected to a water jacket integral to the cuvette holder and monitored by a sensor directly located in the holder. The second derivative of the resulting data was then used to calculate the inflection point of the transition and, hence, the midpoint for the thermal transition.
Acknowledgments
This work was funded by grants from the Australian Research Council, the National Health and Medical Research Council (Australia), and the National Heart Foundation of Australia. S.P.B. is a Logan Fellow of Monash University and J.C.W. is a Peter Doherty Fellow of the National Health and Medical Research Council (Australia) (Grant no. 997144). The authors gratefully acknowledge the donation of time-expired plasma by the Australian Red Cross Blood Service.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.24101.
References
- Beauchamp, N.J., Pike, R.N., Daly, M., Butler, L., Makris, M., Dafforn, T.R., Zhou, A., Fitton, H.L., Preston, F.E., Peake, I.R., and Carrell, R.W. 1998. Antithrombins Wibble and Wobble (T85M/K): Archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood 92 2696–2706. [PubMed] [Google Scholar]
- Bock, S.C., Marrinan, J.A., and Radziejewska, E. 1988. Antithrombin III Utah: Proline-407 to leucine mutation in a highly conserved region near the inhibitor reactive site. Biochemistry 27 6171–6178. [DOI] [PubMed] [Google Scholar]
- Bottomley, S.P. and Stone, S.R. 1998. Protein engineering of chimeric Serpins: An investigation into effects of the serpin scaffold and reactive center loop length. Protein Eng. 11 1243–1247. [DOI] [PubMed] [Google Scholar]
- Carrell, R.W. and Gooptu, B. 1998. Conformational changes and disease—Serpins, prions and Alzheimer's. Curr. Opin. Struct. Biol. 8 799–809. [DOI] [PubMed] [Google Scholar]
- Chang, W.-S., Whisstock, J., Hopkins, P.C., Lesk, A.M., Carrell, R.W., and Wardell, M.R. 1997. Importance of the release of strand 1C to the polymerization mechanism of inhibitory serpins. Protein Sci. 6 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase, T. Jr. and Shaw, E. 1969. Comparison of the esterase activities of trypsin, plasmin, and thrombin on guanidinobenzoate esters. Titration of the enzymes. Biochemistry 8 2212–2224. [DOI] [PubMed] [Google Scholar]
- Dunstone, M.A., Dai, W., Whisstock, J.C., Rossjohn, J., Pike, R.N., Feil, S.C., Le Bonniec, B.F., Parker, M.W., and Bottomley, S.P. 2000. Cleaved antitrypsin polymers at atomic resolution. Protein Sci. 9 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelund, R., Rodenburg, K.W., Andreasen, P.A., Rasmussen, M.S., Guldberg, R.E., and Petersen, T.E. 1998. An ester bond linking a fragment of a serine proteinase to its serpin inhibitor. Biochemistry 37 6375–6379. [DOI] [PubMed] [Google Scholar]
- Egelund, R., Petersen, T.E., and Andreasen, P.A. 2001. A serpin-induced extensive proteolytic susceptibility of urokinase-type plasminogen activator implicates distortion of the proteinase substrate-binding pocket and oxyanion hole in the serpin inhibitory mechanism. Eur. J. Biochem. 268 673–685. [DOI] [PubMed] [Google Scholar]
- Eldering, E., Verpy, E., Roem, D., Meo, T., and Tosi, M. 1995. COOH-terminal substitutions in the serpin C1 inhibitor that cause loop overinsertion and subsequent multimerization. J. Biol. Chem. 270 2579–2587. [DOI] [PubMed] [Google Scholar]
- Elliott, P.R., Lomas, D.A., Carrell, R.W., and Abrahams, J.P. 1996. Inhibitory conformation of the reactive loop of alpha 1-antitrypsin. Nat. Struct. Biol. 3 676–681. [DOI] [PubMed] [Google Scholar]
- Elliott, P.R., Abrahams, J.P., and Lomas, D.A. 1998. Wild-type alpha 1-antitrypsin is in the canonical inhibitory conformation. J. Mol. Biol. 275 419–425. [DOI] [PubMed] [Google Scholar]
- Gettins, P.G.W., Patston, P.A., and Olson, S.T. 1996. Serpins: Structure, function and biology. R.G. Landes Co., Austin, TX.
- Gils, A. and Declerck, P.J. 1998. Structure–function relationships in serpins: Current concepts and controversies. Thromb. Haemost. 80 531–541. [PubMed] [Google Scholar]
- Hood, D.B., Huntington, J.A., and Gettins, P.G. 1994. Alpha 1-proteinase inhibitor variant T345R. Influence of P14 residue on substrate and inhibitory pathways. Biochemistry 33 8538–8547. [DOI] [PubMed] [Google Scholar]
- Hopkins, P.C. and Stone, S.R. 1995. The contribution of the conserved hinge region residues of alpha1-antitrypsin to its reaction with elastase. Biochemistry 34 15872–15879. [DOI] [PubMed] [Google Scholar]
- Hopkins, P.C., Chang, W.-S., Wardell, M.R., and Stone S.R. 1997. Inhibitory mechanism of serpins. Mobility of the C-terminal region of the reactive-site loop. J. Biol. Chem. 272 3905–3909. [DOI] [PubMed] [Google Scholar]
- Huntington, J.A., Pannu, N.S., Hazes, B., Read, R.J., Lomas, D.A., and Carrell, R.W. 1999. A 2.6 Å structure of a serpin polymer and implications for conformational disease. J. Mol. Biol. 293 449–455. [DOI] [PubMed] [Google Scholar]
- Huntington, J.A., Read, R.J., and Carrell, R.W. 2000. Structure of a serpin–protease complex shows inhibition by deformation. Nature 407 923–926. [DOI] [PubMed] [Google Scholar]
- Kaslik, G., Kardos, J., Szabo, E., Szilagyi, L., Zavodszky, P., Westler, W.M., Markley, J.L., and Graf, L. 1997. Effects of serpin binding on the target proteinase: Global stabilization, localized increased structural flexibility, and conserved hydrogen bonding at the active site. Biochemistry 36 5455–5464. [DOI] [PubMed] [Google Scholar]
- Lane, D.A., Olds, R.J., Conard, J., Boisclair, M., Bock, S.C., Hultin, M., Abildgaard, U., Ireland, H., Thompson, E., Sas, G., Horellou, M.H., Tamponi, G., and Thein, S.L. 1992. Pleiotropic effects of antithrombin strand 1C substitution mutations. J. Clin. Invest. 90 2422–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, D.A., Ginsburg, D., Day, D.E., Berkenpas, M.B., Verhamme, I.M., Kvassman, J.O., and Shore, J.D. 1995. Serpin–protease complexes are trapped as stable acyl-enzyme intermediates. J. Biol. Chem. 270 25309–25312. [DOI] [PubMed] [Google Scholar]
- Mottonen, J., Strand, A., Symersky, J., Sweet, R.M., Danley, D.E., Geoghegan, K.F., Gerard, R.D., and Goldsmith, E.J. 1992. Structural basis of latency in plasminogen activator inhibitor-1. Nature 355 270–273. [DOI] [PubMed] [Google Scholar]
- Nakagawa, M., Tanaka, S., Tsuji, H., Takada, O., Uno, M., Hashimoto-Gotoh, T., and Wagatsuma, M. 1991. Congenital antithrombin III deficiency (AT-III Kyoto): Identification of a point mutation altering arginine-406 to methionine behind the reactive site. Thromb. Res. 64 101–108. [DOI] [PubMed] [Google Scholar]
- Potempa, J., Korzus, E., and Travis, J. 1994. The serpin superfamily of proteinase inhibitors: Structure, function, and regulation. J. Biol. Chem. 269 15957–15960. [PubMed] [Google Scholar]
- Schechter, I. and Berger, A. 1967. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27 157–162. [DOI] [PubMed] [Google Scholar]
- Schreuder, H.A., de Boer, B., Dijkema, R., Mulders, J., Theunissen, H.J., Grootenhuis, P.D., and Hol, W.G. 1994. The intact and cleaved human antithrombin III complex as a model for serpin-proteinase interactions. Nat. Struct. Biol. 1 48–54. [DOI] [PubMed] [Google Scholar]
- Shore, J.D., Day, D.E., Francis-Chmura, A.M., Verhamme, I., Kvassman, J., Lawrence, D.A., and Ginsburg, D. 1995. A fluorescent probe study of plasminogen activator inhibitor-1. Evidence for reactive centre loop insertion and its role in the inhibitory mechanism. J. Biol. Chem. 270 5395–5398. [DOI] [PubMed] [Google Scholar]
- Skinner, R., Abrahams, J.P., Whisstock, J.C., Lesk, A.M., Carrell, R.W., and Wardell, M.R. 1997. The 2.6 angstrom structure of antithrombin indicates a conformational change at the heparin binding site. J. Mol. Biol. 266 601–609. [DOI] [PubMed] [Google Scholar]
- Stavridi, E.S., O'Malley, K., Lukacs, C.M., Moore, W.T., Lambris, J.D., Christianson, D.W., Rubin, H., and Cooperman, B.S. 1996. Structural change in alpha-chymotrypsin induced by complexation with alpha 1-antichymotrypsin as seen by enhanced sensitivity to proteolysis. Biochemistry 35 10608–10615. [DOI] [PubMed] [Google Scholar]
- Stein, P.E. and Carrell, R.W. 1995. What do dysfunctional serpins tell us about molecular mobility and disease? Nat. Struct. Biol. 2 96–113. [DOI] [PubMed] [Google Scholar]
- Stone, S.R. and Hofsteenge, J. 1986. Kinetics of the inhibition of thrombin by hirudin. Biochemistry 25 4622–4628. [DOI] [PubMed] [Google Scholar]
- Stone, S.R. and Hermans, J.M. 1995. Inhibitory mechanism of serpins. Interaction of thrombin with antithrombin and protease nexin 1. Biochemistry 34 5164–5172. [DOI] [PubMed] [Google Scholar]
- Stone, S.R. and Le Bonniec, B.F. 1997. Inhibitory mechanism of serpins. Identification of steps involving the active-site serine residue of the protease. J. Mol. Biol. 265 344–362. [DOI] [PubMed] [Google Scholar]
- Stratikos, E. and Gettins, P.G. 1997. Major proteinase movement upon stable serpin–proteinase complex formation. Proc. Natl. Acad. Sci. 94 453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tew, D.J. and Bottomley, S.P. 2001. Intrinsic fluorescence changes and rapid kinetics of proteinase deformation during serpin inhibition. FEBS Lett. 494 30–33. [DOI] [PubMed] [Google Scholar]
- Tucker, H.M., Mottonen, J., Goldsmith, E.J., and Gerard, R.D. 1995. Engineering of plasminogen activator inhibitor-1 to reduce the rate of latency transition. Nat. Struct. Biol. 2 442–445. [DOI] [PubMed] [Google Scholar]
- Verpy, E., Couture-Tosi, E., Eldering, E., Lopez-Trascasa, M., Spath, P., Meo, T., and Tosi, M. 1995. Crucial residues in the carboxy-terminal end of C1 inhibitor revealed by pathogenic mutants impaired in secretion or function. J. Clin. Invest. 95 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisstock, J., Skinner, R., and Lesk, A.M. 1998. An atlas of serpin conformations. Trends Biochem. Sci. 23 63–67. [DOI] [PubMed] [Google Scholar]