

Abstract
We have calculated the stability of decoy structures of several proteins (from the CASP3 models and the Park and Levitt decoy set) relative to the native structures. The calculations were performed with the force field–consistent ES/IS method, in which an implicit solvent (IS) model is used to calculate the average solvation free energy for snapshots from explicit simulations (ESs). The conformational free energy is obtained by adding the internal energy of the solute from the ESs and an entropic term estimated from the covariance positional fluctuation matrix. The set of atomic Born radii and the cavity-surface free energy coefficient used in the implicit model has been optimized to be consistent with the all-atom force field used in the ESs (cedar/gromos with simple point charge (SPC) water model). The decoys are found to have a consistently higher free energy than that of the native structure; the gap between the native structure and the best decoy varies between 10 and 15 kcal/mole, on the order of the free energy difference that typically separates the native state of a protein from the unfolded state. The correlation between the free energy and the extent to which the decoy structures differ from the native (as root mean square deviation) is very weak; hence, the free energy is not an accurate measure for ranking the structurally most native-like structures from among a set of models. Analysis of the energy components shows that stability is attained as a result of three major driving forces: (1) minimum size of the protein-water surface interface; (2) minimum total electrostatic energy, which includes solvent polarization; and (3) minimum protein packing energy. The detailed fit required to optimize the last term may underlie difficulties encountered in recovering the native fold from an approximate decoy or model structure.
Keywords: Protein conformation, free energy, scoring function, ES/IS method, implicit solvation model, molecular surface, electrostatic free energy, internal packing energy
During the past decade, genome sequencing has revealed a vast number of new unknown protein sequences. The growing gap between known sequences and solved structures increases the usefulness and interest in the development of reliable computational methods to predict unknown structures. Recently, we have developed the ES/IS (explicit simulation/implicit solvent) method to estimate free energy of a macromolecule in water solvent based on a strictly statistical-mechanical, physical basis (Vorobjev et al. 1998; Vorobjev and Hermans 1999). The essence of the ES/IS method is a fast but still accurate calculation of the total protein free energy achieved by averaging over snapshots from a short molecular dynamic trajectory in explicit water. Proper statistical averaging over solvent configurations is obtained by using the effective solvation free energy in an IS model, which includes the free energy of molecular cavity formation; an explicit van der Waals solute-solvent interaction; and the free energy of solvent polarization via the continuum dielectric model. It was shown that the ES/IS method with IS model and atomic Born radii (which define the protein-water dielectric surface interface) from the PARSE parameters set (Y.N. Vorobjev and H.A. Hermans, in prep.) was able to correctly discriminate misfolded structures of protein of the EMBL library (Vorobjev et al. 1998). A very similar method (MMPBSA) has been independently developed by Kollman, Case, and coworkers (Srinivasan et al. 1998) and has been applied to estimate the stability of different conformations of proteins and nucleic acids. Recently, we optimized the empirical parameters of the IS model (molecular cavity surface energy and atomic Born radii) to be consistent with experimental data and the underlying molecular mechanics force field (cedar/gromos force field [Hermans et al. 1984] with the simple point charge (SPC) water model [Y.N. Vorobjev and J. Hermans, in prep.]) that is used in the molecular dynamic simulations. The continuum dielectric method with optimized atomic Born radii reproduces a set of 40 microscopically simulated solvent polarization free energies for charged and polar groups of dipeptides with accuracy within 3%, and the same was found to be true for a small protein, eglin (Y.N. Vorobjev and H.A. Hermans, in prep.). The optimization is important to achieve better accuracy of the solvation free energy and a consistency between the potential of mean force of the explicit solvent model and forces of the IS model; that is, the lowest free energy structure of the IS model should have optimal free energy in the ES model. The ES/IS model is also successful in the application to a calculation of a free energy of binding of protein with ligand (Y.N. Vorobjev, unpubl.).
In an earlier study, we found that the ES/IS method was able to consistently distinguish grossly misfolded structures from native structures. The current study considers two new questions. The first is whether the ES/IS method is able to distinguish between the native fold and misfolded structures much more similar to it, and the second is whether a knowledge of the conformational free energy can be useful in guiding the selection of one or more good structures from among models resulting from ab-initio protein folding studies. With these objectives in mind, we have studied a large number (several hundred) of good decoy structures of several small- to middle-sized globular proteins from the decoy library of Park and Levitt and from the CASP3 models (Park and Levitt 1996; see also http://dd.stanford.edu/).
With regard to the first question, we find the ES/IS method gives consistently higher free energies for the decoy structures. With regard to the second question, we find only a weak correlation between the ES/IS free energy and the extent of deformation of the decoy as measured by root mean square deviation (RMSD) of atomic positions, and hence, we conclude that the criterion of low free energy cannot serve to consistently select those decoys most similar to the correct fold.
The strict physical basis of the ES/IS model allows definition of major stability determinants of native protein structure. Analysis leads to the conclusion that protein structural stability (in water) results from simultaneous optimization compactness of the protein-water molecular surface interface, of packing energy, and of electrostatic potential of mean force. The importance of optimization of steric, hydrophobic, and hydrophilic interaction determinants of the globular protein structure stability have been widely recognized empirically (Dill 1990; Vasquez et al. 1994; Honig 1999). The uncovered trend of native protein structures toward optimal total electrostatic free energy (in solvent) provides new insight into the problem of protein structural stability.
The ES/IS calculation of decoy structures probes the multidimensional protein energy surface (folding funnel), relating structural distortion and free energy. When the distortion is projected onto a single coordinate, the RMSD of atomic positions, the correlation with the total free energy of decoys is weak, indicating the existence of an irregular free energy landscape in the neighborhood of the native conformation.
The consistent robustness of the ES/IS free energies allows one to distinguish decoy from native structure, and these free energies can serve as a criterion to select, in the final stages of an ab-initio protein folding algorithm, a few best low free energy decoys from a large number generated via a coarse-grained protein structure prediction method. Although this criterion is relatively weak, recent results for the estimation of decoy structures of similar small proteins via two- and four-body knowledge-based statistical potentials (Bauer and Beyer 1994; Moult 1997; Vajda et al. 1997) show that these (incorrectly) assign a better score to many decoys (Gan et al. 2001).
The ES/IS method
The free energy of a macromolecule in a solvent in a macroscopic conformation A (conformational subspace A) can generally be presented in terms of average energy and entropy over the molecular degrees of freedom (Vorobjev et al. 1998; Vorobjev and Hermans 1999
 |
(1) |
where 〈〉A denotes an average over microconfigurations of the conformation A, Um represents the intraprotein conformational energy, and Sconf,A is the entropy of conformation A. The solvation free energy ΔW(x) is written as a sum of terms for cavity formation, solute-water van der Waals interactions, and electrostatic polarization of solvent by the polar components of the solute. As a result, Equation 1 becomes
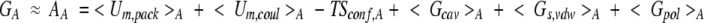 |
(2) |
where the intramolecular potential energy Um has been represented as a sum of short-range energy of packing terms (i.e., for angle deformation and van der Waals interaction), Um,pack; and electrostatic coulombic interactions, Um,coul. A set of microscopic configurations xA,i of a solute in a solvent is generated by molecular dynamics simulation with explicit solvent along a relatively short trajectory, say 50 ps, as snapshots at a fixed time interval (Vorobjev et al. 1998). Of the six terms in Equation 2, three (i.e., 〈Um,pack〉, 〈Um,coul〉, and 〈Gs,vdw〉) are accumulated as averages during the molecular dynamics simulation. The free energy of van der Waals interactions between solute and solvent, Gs,vdw, can be accurately approximated by the potential energy of these interactions, Us,vdw, which can be calculated easily during a molecular dynamics simulation. The explicit calculation of the solute-solvent van der Waals energy is more accurate than the average surface-dependent term of the PARSE model (Sitkoff et al. 1994; Srinivasan et al. 1998).
Simulation protocol
A rigorous estimation of the free energy of a stable conformation of a protein should be taken as a convergent value of Equation 2 as simulation time is increased. Such convergence is not achievable in a free dynamics simulation of a globally unstable conformation. Rather than obtain quasi-stability by application of restraints to atomic coordinates and thereby perturb the internal protein atomic dynamics and atom-atom correlation, we have elected to use a relatively short free dynamic simulation, short enough to exclude structure drift but long enough to achieve steric relaxation and collect a sufficient number of solute-solvent configurations to average over fluctuations of the solute-solvent dielectric interface. We found it adequate to collect 40 to 50 microconformations during a 10-ps trajectory to get reasonable averaging over fast intramolecular atomic fluctuations and minimal conformational drift (RMSD <0.25 Å). The time of 10 ps is ∼4*τc, where τc of ∼2.5 ps is the orientational correlation time of water molecules of the SPC water solvent model, which is in reasonable agreement with experimental data (Svishchev and Kusalik 1994; Levitt et al. 1997).
The following complete protocol of the ES/IS method was used in this paper. Step 1 was the addition of hydrogen atoms to the structure and steric optimization in a vacuum with charges turned off; step 2, solvation of the structure with explicit SPC water to give a solvation volume extending at least 8 Å from any protein atom in all directions. Step 3 was the optimization of the water structure with the protein charges turned off; step 4, six-stage slow heating to a final temperature of 300 K with 3-ps dynamics at each temperature. Step 5 was 5-ps equilibration with all solvent and protein atoms moving, 10 Å cut off on nonbonded interactions, constant bond length with the SHAKE method, and particle mesh Ewald for the long-range electrostatic forces. Step 6 was 10-ps trajectory collection at 300 K and 1 bar, with 0.25-ps snapshots; otherwise it was the same protocol as in step 5. The MD simulations were performed with the Sigma program (Svishchev and Kusalik 1994; Levitt et al. 1997), and the solvation free energies were calculated using the improved Fambe-Sims method (Vorobjev and Hermans 1997; Vorobjev and Scheraga 1997; Vorobjev 1999a,1999b). A completely automated script that exactly realizes the above protocol has been used for each structure in the set. For a protein of 60 to 100 residues, the complete protocol takes 6 to 8 h on a single central processing unit of an SGI-Origin 200 computer with an R10000 processor.
Source of decoy structures
Protein structure decoys have been collected from the results of a blind protein structure prediction contest CASP3 (http://www.ncbi.nlm.nih.gov/Structure/RESEARCH/casp3/index.html) and from Park and Levitt's Stanford protein decoy library (Park and Levitt 1996). The CASP3 structures of proteins 1HKA (T0043 target, 158 residues) and 1BK7 (T0082, 190 residues) have been arbitrarily chosen, taking into account two criteria, that is, the protein should be monomeric and coordinates for all residues should be present in the predicted structures. Several low-RMSD decoys of 1HKA have been obtained by us by refolding the native structure via MD with slow heating to 600 K and then slow cooling to 300 K. Decoys for five small proteins—1ctf, 1r69, 1sn3, 2cro, and 4pti, each with ∼60 residues—have been taken from the Park and Levitt decoy library (Park and Levitt 1996). Of these proteins, all near-native decoys with RMSD (of nonhydrogen atoms, relative to the native structure) <3 Å and roughly half of all decoys with RMSD between 3 Å and 6 Å have been used.
Results
Total free energy
The results of calculations of the total ES/IS free energy of the protein decoy structures according to Equation 2 are shown in Figures 1 and 2 ▶ ▶, as the excess free energy ΔGtot = Gtot (decoy) − Gtot(native) plotted as a function of the RMSD of the averaged structure obtained during the dynamics simulation of the decoy (or native) structure relative to the native X-ray structure. By definition, ΔGtot = 0 for the native structures, but simulation of native protein structures typically produces small non-zero deviations with respect to the starting structure. These are similar for different force fields (Lee et al. 2001a) and reflect both inaccuracies in the atomic force field and differences between the experimental environment and simulation conditions (temperature, buffer, and crystal state versus water box with periodic boundary conditions). It can be seen from Figures 1 and 2 ▶ ▶ that all decoys have higher free energy than do their native structures. The energy gap between the free energy of the best decoys and their respective native structures is between 15 and 20 kcal/mole, that is, the native state is well separated in free energy from a cloud of decoy structures. The correlation coefficients between the excess free energy ΔGtot and RMSD of decoys for different proteins are low, ranging from 0.45 to 0.60.
Fig. 1.

The total excess free energy as a function of the RMSD for decoys of three proteins (1ctf, 1r69, and 4pti) from the Park and Levitt decoy set (Park and Levitt 1996). The excess energy is defined relative to the native structure. The two lowest energy points, RMSD 3.2 Å (circles) and RMSD 3.6 Å (diamonds), belong to the 1ctf_a19727 and 4pti_c20227 decoys, respectively; the dashed line is the minimum discrimination line.
Fig. 2.

The total excess free energy as a function of the RMSD for models of the CASP3 targets t004 (1HKA) and t0082 (1BK7) and the Park and Levitt decoys; the dashed line is the minimum discrimination line.
The minimum discriminatory slope (MDS; Gatchell et al. 2000), defined as the slope of the line that constitutes the lower boundary of the points of the RMSD-versus-energy plots, is equal to 9.1, 17.1, and 17.6 kcal/(mole Å) for the 4pti, 1ctf, and 1r69 decoy sets, respectively (Fig. 1 ▶). The MDS value for the ES/IS free energies is twice as large as that found in calculations of decoy free energy with solvation models based on empirical atomic surface parameters and atomic contact energies (Gatchell et al. 2000).
Energy components
Analyses of different terms of the total free energy of decoys are shown in Figures 3 through 12 ▶ ▶ ▶ ▶ ▶ ▶ ▶ ▶ ▶ ▶. The short-range packing energy, Upack, of Equation 2is consistently optimal for the native structure (Figs. 3, 4 ▶ ▶), as had also been found to be the case for a set of misfolded protein structures (Vorobjev and Hermans 1999). The molecular surface area, which defines a free energy of cavity formation, is minimal for the native structure (Figs. 5, 6 ▶ ▶). As for the sum of intraprotein electrostatic interaction energy and free energy of solvent polarization, Ucoul + Gpol, representing the total free energy of long-range interactions in solvent, it can be seen to be lower for the native structure than for the majority of decoys (Figs. 7, 8 ▶ ▶), with only 1% or 2% of decoys having a lower long-range free energy, but by a margin of <20 kcal/mole.
Fig. 3.

The excess packing energy as a function of the RMSD for the Park and Levitt decoys.
Fig. 4.

The excess packing energy as a function of the RMSD for the CASP3 models and the Park and Levitt decoys.
Fig. 5.

The excess molecular surface area as a function of the RMSD for the Park and Levitt decoys.
Fig. 6.

The excess molecular surface area as a function of the RMSD for the CASP3 models and the Park and Levitt decoys.
Fig. 7.

The excess total electrostatic free energy as a function of the RMSD for the Park and Levitt decoys.
Fig. 8.

The excess total electrostatic free energy as a function of the RMSD for the CASP3 models and the Park and Levitt decoys.
Fig. 9.

The excess internal electrostatic energy as a function of the RMSD for the Park and Levitt decoys.
Fig. 10.

The excess internal electrostatic energy as a function of the RMSD for the CASP3 models and the Park and Levitt decoys.
Fig. 11.

The excess total energy in a vacuum as a function of the RMSD for the Park and Levitt decoys.
Fig. 12.

The excess total energy in a vacuum as a function of the RMSD for the CASP3 models and the Park and Levitt decoys.
In contrast, the internal electrostatic energy Ucoul is not optimal for the native structure. More than 20% of decoy structures have the electrostatic energy lower by several hundreds of kcal/mole than that of the native one (Figs. 9, 10 ▶ ▶). However, for some proteins, for example, 4pti (BPTI), the internal electrostatic energy is optimal for the native structure (Fig. 9 ▶). Because the internal electrostatic energy is not generally optimal for the native structure, the total energy of decoys in vacuum (total internal potential energy, Uvac = Upack + Ucoul) is not generally optimal for the native structures (Figs. 11, 12 ▶ ▶), except rarely, as for BPTI. The strong anticorrelation between the internal electrostatic energy and the solvent polarization free energy is shown in Figure 13 ▶.
Fig. 13.

The excess free energy of solvent polarization versus the internal electrostatic energy for the CASP3 models and the Park and Levitt decoys.
The quasi-harmonic conformational entropy term in Equation 2was found to have a similar value for decoy and native structures (varying by <2 kcal/mole), as had also been found to be true for the misfolded proteins (Vorobjev et al. 1998). Because inclusion of the entropy term has a negligible effect on the free energy differences of a decoy set, we have not included the conformational entropy term in the total free energies reported.
MD relaxation of the low energy decoys
Because we have found decoy structures to be frequently unstable during dynamics simulation, the reported free energies are based on rather short (10 ps long) trajectories of free dynamics simulation, during which the drift of any decoy structure is small. To check if the low energy decoys can easily relax to more native-like structures, we have run 2.5- to 3.0-ns free molecular dynamics for decoys 1ctf_a19727 and 4pti_c20227 (Figs. 14, 15 ▶ ▶). One sees that the free energy of one of these decoys decreases slightly, with little change of the RMSD, whereas that of the other remains constant, with a slight increase of the RMSD. Thus, there is no evidence for the occurrence of a trend toward the native structure on this rather short timescale.
Fig. 14.

Rmsd versus simulation time in a free molecular dynamic relaxation of two low-energy decoys (4pti_c20227 and 1ctf_a19727) from the Park and Levitt decoy set.
Fig. 15.

The total free energy versus simulation time in a free molecular dynamic relaxation of two low-energy decoys (Fig. 14 ▶); the thin lines represent instantaneous values; the bold lines, averages over a 50-ps window.
Discussion
Energetics of the native state
The optimality of the native structure in water solvent results from a delicate balance between the internal electrostatic energy Ucoul and the solvent polarization Gpol energy, which are strongly coupled (Fig. 13 ▶). The high anticorrelation between the internal electrostatic energy and free energy of solvent polarization has been previously noticed (Vorobjev and Hermans 1999). In the present study, we find average correlation coefficients of Ucoul and Gpol between −0.93 and −0.98, with both energy terms having a similar range of variation, which is of the order of ±1000 kcal/mole for a protein of ∼100 residues. The large range of variation places a strong requirement on the accuracy of the method used for calculation of the free energy of solvent polarization. This makes it important to use a maximally accurate method to compute the free energy of solvation and optimize the compensation between intrasolute and solute-solvent electrostatic free energy.
Apparently, the stability of native protein structures is a result of simultaneous optimization of three energy terms: (1) atom packing energy as the energy of all short-range interactions (van der Waals, torsion, and deformation energy), (2) the energy of hydrophobic cavity formation, and (3) total electrostatic free energy (including the free energy of interaction of protein charges with solvent). Analysis of the free energy terms that favor the native structure confirms the importance of hydrophobic and hydrophilic interactions with solvent (Dill 1990) as dominant forces determining stability of globular proteins. In addition, the importance of good atomic packing is strikingly emphasized. In that context, it is worth noting that a recent analysis of high-resolution crystal structures of proteins has revealed that the "bump-into-hole" model of what constitutes a good fit extends even to packing at the level of the hydrogen atoms of the protein (Word et al. 1999).
Alternative ways of scoring protein conformation
The ES/IS method (Vorobjev et al. 1998; Vorobjev and Hermans 1999) is based strictly on physical models and is found to discriminate the native protein structure as the structure with minimal free energy in water against both major and minor changes in structure, the native structure being separated from the best decoys by a free energy gap of between 15 and 20 kcal/mole. Recently reported calculations of the free energy (Lazaridis and Karplus 1999a; Gatchell et al. 2000) of the Park and Levitt decoy set make use of a simple empirical solvation model based on atomic contributions calibrated for a model set of small molecule compounds and on knowledge-based contact potentials, with electrically neutral representation of charged side-chains (Arg, Lys, Asp, and Glu), a dielectric constant dependent on interatomic distance, r, taken as r/Å (Lazaridis and Karplus 1999a) or 4r/Å plus a special protocol for van der Waals normalization (Gatchell et al. 2000). A recent assessment of decoy structures of similar small proteins with two- and four-body knowledge-based statistical potentials (Gan et al. 2001) provides a lower score for many decoys. The ES/IS method shows twice the discrimination power of the best simplified method (Gan et al. 2001).
The use of more approximate IS models (Lazaridis and Karplus 1999b; Roux and Simonson 1999; Dominy and Brooks 2001) is justified when the objective is to assess numerous decoys in the context of optimizing the structure of a protein when structural information is incomplete or absent; however, speedup is achieved at the cost of departing from a strictly physics-based potential. The quality of the more approximate solvation models should be evaluated by a comparison with the results of more accurate models, with the ES/IS method and related MMPBSA method (Srinivasan et al. 1998) arguably achieving the greater accuracy.
Protein folding and simulations of protein folding
Although we have found only a weak correlation between conformational free energy and distance of the structure from the native, as measured by the RMSD, our results also indicate that the free energy of the best decoys systematically increases with an increase of the RMSD (Figs. 1, 2 ▶ ▶). These results indicate that the energy surface in the neighborhood of the native conformation is rough, with the lowest minima forming a funnel toward the native state. This, together with the observation of a free energy gap of between 15 and 20 kcal/mole, agrees well with current views (Honig 1999; Dinner et al. 2000). In fact, the free energy gap is of the same order as the amount by which the native state of proteins is typically stabilized relative to the unfolded or denatured state, and it ensures that misfolded conformations are not stable or only marginally stable relative to the unfolded state.
Although the conformational free energy estimate provided by the ES/IS method correlates only weakly with the extent to which the decoy structures deviate from the native state, this free energy is able to distinguish between native state and non-native decoys. Current approaches to the problem of determining the conformation of a protein on the basis of the amino acid sequence use a variety of scoring functions to assess very large numbers of conformations (CASP3, see http://www.ncbi.nlm.nih.gov/Structure/RESEARCH/casp3/index.html). Evaluation of such a scoring function must be very fast, and this is achieved by simplifications that tend to lower the achievable accuracy, with the correct fold not having the lowest score. Therefore, such a method should be used to generate a large set of low-scoring structures to increase the likelihood that the correct fold will be included. This large set can then be scored with a more accurate function, if necessary in stages, with use of ES/IS (or MMPBSA; Srinivasan et al. 1998) as a final step. A recently created library containing a large number of decoys generated by ab-initio structure prediction (Simons et al. 1999; Baker 2000) offers a test bed for development of a complete protocol. A study by Baker, Kollman, and coworkers uses the MMPBSA method with such a protocol, apparently with considerable success (Lee et al. 2001b).
The fact that different energy terms are found to simultaneously be optimal for the native structure does not mean that any one of these terms alone can be isolated as the driving force in model calculations aimed at determining the most stable protein conformation. For example, it has been shown that optimization of residue contact potentials based on observed frequencies of residue contacts in proteins of known structure leads to structures in which the extent of hydrophobic contacts is excessive (Zhang 1999). Our results clearly indicate that an assumption in folding simulations that the electrostatic interactions in vacuum are optimal (as in the electrostatically driven Monte Carlo global optimization method [Ripoll and Scheraga 1988]) cannot be widely applicable but should be replaced by the assumption that a total electrostatic free energy that includes solvent polarization is optimal. (Evaluation of the latter term may, of course, be prohibitively slow.) Use of an optimization method that attempts to fit the structure into a predefined compact cavity (Liwo et al. 1993) obviously has a better chance of success. A major difficulty in constructing a workable algorithm for protein folding simulation lies in the need to combine the three components (i.e., global electrostatics, compactness, and close fit) correctly and in the right proportion, in a very rapidly evaluated approximation.
We are struck by how the native structures consistently show a significantly lower packing energy than that of the decoys. This finding is consistent with earlier findings that the interior space of the proteins is used efficiently (Richards 1977) and with the result of a recent analysis of high-resolution protein structures indicating that an interdigitating arrangement is common (also) at the level of the hydrogen atoms (Word et al. 1999). This finding leads us to make two suggestions. First, the favorable energy derived from a very detailed final fit is an important factor that causes folding in a unique conformation rather than to a less ordered molten globule state. Second, the precise fit will be hard to find in folding simulations, both because for the sake of computational efficiency these use reduced models, and because a small offset between contacting faces will destroy the fit. Although progress in folding simulations has focused on the generation of models that globally resemble their native structures, the problem of refining a globally correct but locally incorrect model to produce a complete native-like structure adds another major level of complexity.
Acknowledgments
This work was supported by a research grant from the National Center for Research Resources, National Institutes of Health (RR08012).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.15501.
References
- Baker, D.A. 2000. The Baker Laboratory homepage. http://depts.washington.edu/bakerpg/
- Bauer, A. and Beyer, A. 1994. An improved pair potential to recognize native protein folds. Proteins 18 254–261. [DOI] [PubMed] [Google Scholar]
- Dill, K.A. 1990. Dominant forces in protein folding. Biochemistry 29 7133–7155. [DOI] [PubMed] [Google Scholar]
- Dinner, A.R., Sali, A., Smith, L.J., Dobson, C.M., and Karplus, M. 2000. Understanding protein folding via free-energy surfaces from theory and experiment. Trends Biochem. Sci. 25 331–339. [DOI] [PubMed] [Google Scholar]
- Dominy, B.N., and Brooks, C.L. 2001. Identifying native-like protein structures using physics-based potentials. J. Comp. Chem. (in press). [DOI] [PubMed]
- Gan, H.H., Tropsha, A., and Schlick, T. 2001. Lattice protein folding with two and four-body statistical potentials. Proteins 43 161–174. [DOI] [PubMed] [Google Scholar]
- Gatchell, D.W., Sheldon, D., and Vajda, S. 2000. Discrimination of the near-native protein structures from misfolded models by empirical free energy functions. Proteins 41 518–534. [PubMed] [Google Scholar]
- Hermans, J., Berendsen, H.J.C., van Gunsteren, W.F., and Postma, J.P.M. 1984. A consistent empirical potential for water-protein interactions. Biopolymers 23 1513–1518. [Google Scholar]
- Honig, B. 1999. Protein folding: From the Levinthal paradox to structure predictions. J. Mol. Biol. 293 283–293. [DOI] [PubMed] [Google Scholar]
- Lazaridis, T. and Karplus, M. 1999a. Discrimination of the native from misfolded protein models with an energy function including implicit solvation. J. Mol. Biol. 288 477–487. [DOI] [PubMed] [Google Scholar]
- ———. 1999b. Effective energy function for protein in solution. Proteins 35 133–152. [DOI] [PubMed] [Google Scholar]
- Lee, M.R., Baker, D., and Kollman, P.A. 2001a. 2.1 and 1.8 Å average Cα RMSD structure predictions on two small proteins, HP-36 and S15. J. Am. Chem. Soc. 123 1040–1046. [DOI] [PubMed] [Google Scholar]
- Lee, M.R., Tsai, J., Baker, D., and Kollman, P. 2001b. Molecular dynamics in the endgame of protein structure prediction. J. Mol. Biol. (in press). [DOI] [PubMed]
- Levitt, M., Hirshberg, M., Sharon, R., Laidig, K.E., and Daggett, V. 1997. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J. Phys. Chem. 101 5051–5061. [Google Scholar]
- Liwo, A., Pincus, M.R., Wawak, R.J., Rackovsky, S., and Scheraga, H.A. 1993. Prediction of protein structure on the basis of search for compact structures: Test on avian pancreatic peptide. Protein Sci. 2 1715–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult, J. 1997. Comparison of database potentials and molecular mechanics force fields. Curr. Opin. Struct. Biol. 7 194–199. [DOI] [PubMed] [Google Scholar]
- Park, B.H. and Levitt, M. 1996. Decoys of globular proteins. J. Mol. Biol. 258 367–392. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. 1977. Areas, volumes, packing, and protein structures. Annu. Rev. Biophys. Bioeng. 6 151–176. [DOI] [PubMed] [Google Scholar]
- Ripoll, D. and Scheraga, H.A. 1988. On the multiple-minima problem in the conformational analysis of polypeptides: An electrostatically driven Monte Carlo method. Biopolymers 27 1283–1303. [DOI] [PubMed] [Google Scholar]
- Roux, B. and Simonson, T. 1999. Implicit solvent models. Biophys. Chem. 78 1–20. [DOI] [PubMed] [Google Scholar]
- Simons, K.T., Bonneau, R., Ruczinski, I., and Baker, D. 1999. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins 37 171–176. [DOI] [PubMed] [Google Scholar]
- Sitkoff, D., Sharp, K.A., and Honig, B. 1994. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98 1978–1988. [Google Scholar]
- Srinivasan, J., Cheatham, T.E., Cieplak, P., Kollman, P.A., and Case, D.A. 1998. Continuum solvent studies of stability of DNA, RNA and phosphoramide DNA helices. J. Am. Chem. Soc. 120 9401–9409. [Google Scholar]
- Svishchev, I.M. and Kusalik, P.G. 1994. Simulation of the dynamic properties of liquid water. J. Phys. Chem. 98 728–736. [Google Scholar]
- Vajda, S., Sippl, M., and Novotny, J. 1997. Empirical potentials and functions for protein folding and binding. Curr. Opin. Struct. Biol. 7 222–228. [DOI] [PubMed] [Google Scholar]
- Vasquez, M., Nemethy, G., and Scheraga, H.A. 1994. Conformational energy calculation on polypeptides and proteins. Chem. Rev. 94 2183–2239. [Google Scholar]
- Vorobjev, Y.N. 1999a. FAMBE: Fast adaptive multigrid boundary element method for macromolecular electrostatics. http://femto.med.unc.edu/FAMBE
- Vorobjev, Y.N. 1999b. SIMS general program for calculation smooth invariant molecular surface. http://femto.med.unc.edu/SIMS
- Vorobjev, Y. and Hermans, J. 1997. SIMS: Computation of a smooth invariant molecular surface. Biophys. J. 73 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1999. ES/IS: Estimation of conformational free energy by combining dynamics simulations with explicit solvent with an implicit solvent continuum model. Biophys. Chem. 78 195–205. [DOI] [PubMed] [Google Scholar]
- Vorobjev, Y.N. and Scheraga, H.A. 1997. A fast adaptive multigrid boundary element method for macromolecular electrostatics in a solvent. J. Comp. Chem. 18 569–583. [Google Scholar]
- Vorobjev, Y.N., Almagro, J.C., and Hermans, J. 1998. Discrimination between native and intentionally misfolded conformations of proteins: ES/IS, a new method for calculating conformational free energy that uses both dynamic simulations with explicit solvent and an implicit solvent continuum model. Proteins 32 399–413. [PubMed] [Google Scholar]
- Word, J.M., Lovell, S.C., LaBean, T.H., Taylor, H.C., Zalis, M.E., Presley, P.K., Richardson, J.S., and Richardson, D.C. 1999. Visualizing and quantifying molecular goodness-of-fit: Small-probe contact dots with explicit hydrogen atoms. J. Mol. Biol. 285 1711–1733. [DOI] [PubMed] [Google Scholar]
- Zhang, H. 1999. A new hybrid Monte Carlo algorithm for protein function test and structure refinement. Proteins 34 464–471. [PubMed] [Google Scholar]