

Abstract
Conventional fold recognition techniques rely mainly on the analysis of the entire sequence of a protein. We present an MBA method to improve performance of any conventional sequence-based fold assignment. The method uses sequence motifs, such as those defined in the Prosite database, and the SwissProt annotation of the fold library. When combined with a simple SDP method, the coverage of MBA is comparable to the results obtained with PSI-BLAST. However, the set of the MBA predictions is significantly different from that of PSI-BLAST, leading to a 40% increase of the coverage for the combined MBA/PSI-BLAST method. The MBA approach can be easily adopted to include the results of sequence-independent function prediction methods and alternative motif and annotation databases. The method is available through the web server localized at http://www.doe-mbi.ucla.edu/mba.
Keywords: Bioinformatics, sequence motif, functional annotation, fold assignment
The Human Genome Project and satellite projects have elucidated the genomic sequences of nearly 100 organisms, including human (see TIGR: http://www.tigr.org/tdb/, National Center for Biotechnology Information [NCBI]: http://www.ncbi.nlm.nih.gov/Genomes/). To fully use this vast amount of information, the raw sequences first have to be processed to identify functional genes. Next, the structures and functions of the proteins they encode must be determined in order to gain insight into the roles of the genes within the living organism.
The rate of the experimental determination of protein structures, although continuously increasing, still lags behind protein sequences by roughly two orders of magnitude. To fill this gap, investigators have developed methods for protein structure and function prediction (for reviews, see Smith 1999; Domingues et al. 2000a; Skolnick and Fetrow 2000; Skolnick et al. 2000). These methods rely almost exclusively on identification of sequence similarity to proteins of known fold or on the compatibility of the new sequence with the chemical environments of individual residues when threaded through previously determined experimental structures.
Sequence-based methods of fold assignment attempt to identify pairs of homologous proteins–proteins that share, because of common ancestry, similar structure and function (Fig. 1 ▶). Dynamic programming-based sequence alignment methods (Needleman and Wunsch 1970; Smith and Waterman 1981) are able to identify homologs when sequence identity is larger then roughly 20%–30%. The use of multiple sequence alignment-based sequence profiles (Gribskov et al. 1987; Altschul et al. 1997) and HMM methods (Karplus et al. 1998) can, at least in some cases, extend the sensitivity of the fold assignments below 20% of sequence identity. Structure-based predictions take into consideration residue preferences for different environments within the structure (Bowie et al. 1991; Jones et al. 1992; Jones 1999; Bienkowska et al. 2000). When combined with the prediction of secondary structure, they can perform about as well as the sequence-based methods (Fischer and Eisenberg 1996; Russell et al. 1996; Jones et al. 1999; Panchenko et al. 2000). Another set of methods rely on the intergenome distribution of homologous proteins to infer their function directly (Marcotte 2000). Those methods, although bypassing the structure-determination step, are able to assign a function to the protein by identifying groups of nonhomologous proteins that coevolved together and thus fulfill similar roles within a cell (Andrade et al. 1997).
Fig. 1.

Flowchart of the MBA method. Conventional fold assignment methods (solid lines) compare the entire sequence (or multiple sequence alignment) of the probe to the sequences (or multiple sequence alignments) or structures of the folds in a fold library. MBA (dashed lines) uses information present in the occurrences of motifs in the probe sequence and target annotation to combine it with a conventional sequence/structure score.
The information about sequence and 3D structure is only a small part of the vast experimental knowledge about proteins. Publicly accessible databases also contain information about protein function, including expression patterns, enzymatic activities and positions within metabolic and signaling pathways, and interactions with other proteins and small molecules (Baxevanis 2000 and references therein). However, until recently, conventional, automated prediction algorithms ignored most of these additional information sources, although they can be used to improve the reliability of prediction. In fact, identification of sequence motifs coupled with the analysis of the functional information about the protein of interest by human experts is one of the approaches most widely used to characterize newly identified proteins.
Functional information was used recently in the later stages of the mostly manual structure prediction of CASP3 targets by Murzin and Bateman (1997) or to identify a possible function of the new protein after initial structure prediction (Zhang et al. 1999). On the other hand, the SAWTED algorithm (MacCallum et al. 2000) allows automatic screening of the potential predicted structures against the functional information about the unknown protein.
The fully automated approach presented here combines the functional information contained in the SwissProt keyword annotation with the Prosite motif database to improve the performance of any conventional sequence- or structure-based prediction. As opposed to the SAWTED approach (MacCallum et al. 2000), our method does not rely on the annotation used to characterize newly identified proteins of the unknown sequence and thus is well suited to analysis of poorly characterized sequences such as those produced by the full-genome sequencing projects.
Results
Conventional fold assignment methods use information present in the entire sequence (or multiple sequence alignment) of the unknown protein (probe). They analyze compatibility of the probe sequence with the sequences and/or structures of the folds present in the fold library to identify the closest structural match (Fig. 1 ▶, left branch). The MBA method presented here (Fig. 1 ▶, right branch) concentrates on the occurrences of common sequence motifs within the probe and target annotation to generate a motif-fold compatibility score. This score is then combined with the sequence-based information to obtain the final MBA score.
Motif–fold compatibility
It has been long known that some regions within protein sequence are crucial for function and thus better conserved among homologs than are surrounding regions (Bork and Koonin 1996; Kasuya and Thornton, 1999). This observation has led to the creation of motif libraries such as Prosite (Hofmann et al. 1999), which catalog patterns repeatedly recurring in protein sequences. The motifs present in the library can be classified as belonging to one of the two groups. Some of them correspond to structural elements, such as coiled-coil and zinc-finger motifs, that are shared by all representatives of a given fold or group of folds. Often, conservation of such motifs is required for proper folding of the protein. The other group of sequence motifs reflects the functions of the molecule: cofactor and ligand binding pockets, catalytic sites, or motifs responsible for interaction with other proteins directly or after posttranslational modification. Usually proteins of similar structure perform similar functions within the cell, and thus it can be expected that the occurrence of not only structural but also of functional motifs would correlate with protein fold, although there are marked exceptions to this expectation (Hegyi and Gerstein 1999).
The correlation between motif presence and protein structure can be evaluated by calculating the log-odds score, SFM, defined as
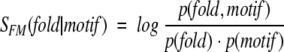 |
(1) |
where p(motif) and p(fold) are probabilities of finding a particular sequence motif and a particular fold in all contiguous CATH domains (Orengo et al. 1999) that are identified in the nonredundant fold library on the basis of the PDB_SELECT list (Hobohm and Sander 1994), and p(fold, motif) is the corresponding joint probability.
Figure 2a ▶ (solid circles) shows the distribution of SFM scores for the folds of our library. The presence of fold–motif pairs characterized by SFM ≫ 1 demonstrates that, indeed, in a number of cases, protein fold is strongly correlated with the presence of particular Prosite motifs. However, within the range −2SFM < 2 there are a large number of uncorrelated pairs. Inspection shows that they are mostly due to the presence of short, weakly defined motifs, such as phosphorylation and myristoylation sites.
Fig. 2.

(a) Frequency of the SFM scores for all continuous protein domains defined in the CATH database (Orengo et al. 1999) and also found in the SwissProt database. Notice that annotation filtering removes a large number of uncorrelated domain–motif pairs having scores SFM ≈ 0. Unfiltered (solid circles) and annotation-filtered (open squares, open circles) motif–fold pairs using CMK = 0.25 and 1.0, respectively. (b) Frequency of the SMK scores for all protein sequences present in the SwissProt database (Bairoch and Apweiler 2000; release 39, 80,000 sequences). Notice that, apart from the vast majority of the uncorrelated motif–keyword pairs, there is also a small subset of strongly correlated pairs for which SMK ≫ 0. It constitutes ∼10%–15% of the total number of sequence motif–keyword pairs.
Table 1 lists the top-scoring fold–motif pairs. The majority of them involve relatively long motifs participating in cofactor or substrate binding. In some cases, the motifs are related to the characteristic structural features of the fold, such as cysteine residues participating in disulfide bridge formation or residues binding ligands such as zinc ions, which stabilize protein structure.
Table 1.
Top-ranking fold–motif pairs by SFMscores
| Motifb | |||
| SFM | Folda [Class:Arch:Topo] | Accession number | ID |
| 7.74 | 4:10:400 | PS01209 | LDLRA_1 |
| 7.74 | 2:102:10 | PS00199/PS00200 | RIESKE_1 |
| RIESKE_2 | |||
| 7.74 | 1:10:610 | PS00592/PS00698 | GLYCOSYL_HYDROL_F9_1 |
| GLYCOSYL_HYDROL_F9_2 | |||
| 7.74 | 1:10:575 | PS00384 | PROKAR_ZN_DEPEND_PLPC |
| 7.33 | 3:90:230 | PS00680/PS01202 | MAP_1 |
| MAP_2 | |||
| 7.33 | 3:50:12 | PS00888/PS00889 | CNMP_BINDING_1 |
| CNMP_BINDING_2 | |||
| 7.33 | 3:40:140 | PS00903 | CYT_DCMP_DEAMINASES |
| 7.33 | 3:30:40 | PS00518 | ZINC_FINGER_C3HC4 |
| 7.33 | 2:60:130 | PS00083 | INTRADIOL_DIOXYGENAS |
| 7.33 | 2:160:10 | PS00101 | HEXAPEP_TRANSFERASES |
| 7.33 | 1:10:340 | PS01155 | ENDONUCLEASE_III_2 |
| 7.04 | 3:50:11 | PS00859/PS00860 | GTP_CYCLOHYDROL_1_1 |
| GTP_CYCLOHYDROL_1_2 | |||
| 7.04 | 3:30:460 | PS00522 | DNA_POLYMERASE_X |
| 7.04 | 3:20:10 | PS00770 | AA_TRANSFER_CLASS_4 |
| 7.04 | 3:10:180 | PS00082 | EXTRADIOL_DIOXYGENAS |
| 7.04 | 2:110:10 | PS00024 | HEMOPEXIN |
| 7.04 | 1:10:230 | PS00480 | CITRATE_SYNTHASE |
| 7.04 | 1:10:120 | PS00426 | CEREAL_TRYP_AMYL_INH |
| 6.82 | 4:10:220 | PS00036 | BZIP_BASIC |
| 6.82 | 4:10:220 | PS00968 | ANTENNA_COMP_ALPHA |
| 6.82 | 3:40:250 | PS00380/PS00683 | RHODANESE_1 |
| RHODANESE_2 | |||
| 6.82 | 2:70:97 | PS01164 | COPPER_AMINE_OXID_1 |
| 6.82 | 2:40:40 | PS00771/PS00772 | BARWIN_1 |
| BARWIN_2 | |||
| 6.82 | 2:40:40 | PS00932 | MOLYBDOPTERIN_PROK_3 |
| 6.82 | 1:20:140 | PS00073 | ACYL_COA_DH_2 |
| 6.64 | 4:10:240 | PS00463 | ZN2_CY6_FUNGAL_1 |
| 6.64 | 3:90:226 | PS00166 | ENOYL_COA_HYDRATASE |
| 6.64 | 3:90:226 | PS00382 | CLP_PROTEASE_HIS |
| 6.64 | 3:90:226 | PS00381 | CLP_PROTEASE_SER |
| 6.64 | 2:160:20 | PS00502 | POLYGALACTURONASE |
a Fold, as specified by the first three positions of the numerical identifier from the CATH classification.
b Accession number and identifier of the sequence motif, as specified in the Prosite database. Only motifs present in at least two different superfamilies are listed.
SFM scores can be used to evaluate the compatibility of a sequence containing a set of motifs with different folds and thus can constitute a basis for a fold assignment method. The contribution of the motifs that are uncorrelated with the protein structure can by eliminated by accepting SFM scores only above a preset cutoff (CFM). The coverage–accuracy curve parameterized by CFM shown in Figure 3 ▶ (solid squares) demonstrates that, in the standardized benchmark used throughout this paper (see Materials and Methods), this simple motif-based prediction assigns folds about as well as a commonly used sequence-based method of Fisher and Eisenberg (1996; crosshairs).
Fig. 3.

Performance of the MBA method, showing both accuracy of the assignment and the percentage of the coverage of the test set of CATH domains (see Materials and Methods), as compared with the SDP method (Fischer and Eisenberg 1996). Notice that the annotation-filtered version of MBA performs at least as well as the SDP method. The performance of the MBA method is parametrized by CFM, (solid squares) and CMK (open squares). Performance of the SDP method parametrized by Z score (+) is shown as a reference.
Keyword filtering
The motifs compiled in the Prosite database were often defined as a set of residues performing a protein's function. For example, they are composed by the residues that form the active site of an enzyme or bind ligands. Some of the motifs are characteristic of structural features such as coiled coils or zinc fingers. For both cases, we might expect that the presence of a given motif correlates with the annotation of a given protein, for example, such as that provided by the keyword field present in SwissProt database records. This correlation can be evaluated in the same way as the motif–fold correlation by the SMK score defined as
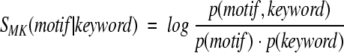 |
(2) |
where p(motif) and p(keyword) are probabilities of finding the motif and keyword in a SwissProt entry, and p(motif, keyword) is the corresponding joint probability. Figure 2b ▶ demonstrates that the distribution of SMK is analogous to that shown for SFM in Figure 2a ▶: apart from the majority of the uncorrelated motif–keyword pairs, there is also a group of motif–keyword pairs that preferentially occur together. As shown in Table 2, most often the pairs involve a sequence motif that is the manifestation of a structural feature or function described by a given keyword. Interestingly enough, high SFM scores (SFM ≫ 0) are observed for specific functional sites even when they are paired with generic keywords describing protein function in a coarse way (e.g., "glycolate pathway," "iron storage," or "folate biosynthesis").
Table 2.
Top-ranking motif–keyword pairs by SMKscores
| Motifa | |||
| SMK | Accession number | ID | Keywordb |
| 8.78 | PS00195 | GLUTAREDOXIN | Deoxyribonucleotide synthesis |
| 8.74 | PS00271 | THIONIN | Thionin |
| 8.70 | PS00271 | THIONIN | Plant toxin |
| 8.46 | PS00630 | IMP_2 | Lithium |
| 8.25 | PS00771/PS00772 | BARWIN_1 | Latex |
| BARWIN_2 | |||
| 8.12 | PS00144 | ASN_GLN_ASE_1 | Aspartic protease inhibitor |
| 7.98 | PS00909 | MR_MLE_2 | Mandelate pathway |
| 7.97 | PS00838 | INTERLEUKIN_4_13 | B-cell activation |
| 7.95 | PS00665/PS00666 | DHDPS_1/DHDPS_2 | Feedback-inhibition |
| 7.85 | PS00549 | BACTERIOFERRITIN | Iron storage |
| 7.83 | PS00557 | FMN_HYDROXY_ACID_DH | Glycolate pathway |
| 7.81 | PS00859/PS00860 | GTP_CYCLOHYDROL_1_1 | Tetrahydrobiopterin biosynthesis |
| GTP_CYCLOHYDROL_1_2 | |||
| 7.80 | PS00289 | PENTAXIN | Pentaxin |
| 7.73 | PS00204 | FERRITIN_2 | Iron storage |
| 7.71 | PS00309 | LECTIN_GALACTOSIDE | Galaptin |
| 7.69 | PS01164 | COPPER_AMINE_OXID_1 | Topaquinone |
| 7.69 | PS00540 | FERRITIN_1 | Iron storage |
| 7.64 | PS01165 | COPPER_AMINE_OXID_2 | Topaquinone |
| 7.56 | PS00485 | A-DEAMINASE | Hereditary hemolytic anemia |
| 7.55 | PS00768/PS00769 | TRANSTHYRETIN_1 | Thyroid hormone |
| TRANSTHYRETIN_2 | |||
| 7.53 | PS01065 | ETF_BETA | Glutaricaciduria |
| 7.50 | PS00506/PS00679 | BETA_AMYLASE_1 | Polysaccharide degradation |
| BETA_AMYLASE_2 | |||
| 7.49 | PS00846 | HTH_ARSR_FAMILY | Cadmium resistance |
| 7.46 | PS00424 | INTERLEUKIN_2 | Immune response |
| 7.45 | PS00557 | FMN_HYDROXY_ACID_DH | Mandelate pathway |
| 7.44 | PS00451 | PATHOGENESIS_BETVI | Pathogenesis-related protein |
| 7.43 | PS00969 | ANTENNA_COMP_BETA | Antenna complex |
| 7.43 | PS00968 | ANTENNA_COMP_ALPHA | Antenna complex |
| 7.43 | PS00494 | BACTERIAL_LUCIFERASE | Photoprotein |
| 7.40 | PS00253 | INTERLEUKIN_1 | Pyrogen |
| 7.39 | PS00792/PS00793 | DHPS_1 | Folate biosynthesis |
| DHPS_2 | |||
| 7.35 | PS00969 | ANTENNA_COMP_BETA | Bacteriochlorophyll |
| 7.35 | PS00968 | ANTENNA_COMP_ALPHA | Bacteriochlorophyll |
| 7.27 | PS00577 | AVIDIN | Biotin |
| 7.25 | PS00768/PS00769 | TRANSTHYRETIN_1 | Polyneuropathy |
| TRANSTHYRETIN_2 | |||
a Accession number and identifier of the motif, as specified in the Prosite database.
b Keyword, as specified by the KW field in the SwissProt entry.
The motif–keyword correlation can be used as an alternative way of selecting the motifs contributing to the fold compatibility score. As can be seen in Figure 2a ▶ (open circles), removal of the fold–motif pairs, for which
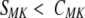 |
(3) |
results in nearly complete elimination of the uncorrelated SFM pairs from the histogram. However, because this type of filtering removes the requirement that the motif is found in every representative of a fold class, correlations for the entire range of the SFM distribution are reduced. In some cases, such lack of the dependence on the strict motif–fold correlation can be advantageous because it permits identification of motifs that are characteristic of a specific fold but absent from some members of the fold class (Orengo et al. 1994). For example, in the case of TIM barrels, there are >50 Prosite motifs characteristic of the functions performed by the representatives of this functionally diverse fold; however, none of the motifs is encountered in all of them.
These keyword-filtered SFM scores can be used to evaluate sequence–fold compatibility in the same manner as the unfiltered scores. As shown in Figure 3 ▶ (open squares), the modified scoring scheme performs better than the initial SFM cutoff-based approach. Notice that annotation of only the target domains was used. A more advanced version of the method could also use annotation of the probe sequence obtained through automated literature scanning or by function prediction. However, to demonstrate that prior knowledge of the probe structure does not affect the performance of the method in the test set, no annotation of the probe sequence was used here.
In addition to improved overall performance, the keyword-filtered version returns a large number of correct fold assignments that are missed by a sequence-only based method, such as PSI-BLAST (Fig. 4 ▶). In fact, for both methods of motif selection, the set of correct motif-based assignments, although comparable in number with the PSI-BLAST results, differs from it by more than 40%. This observation suggests that the motif-based method relies on a different set of information embedded in the protein sequence than the conventional sequence-based methods and thus a combination of both approaches might be beneficial.
Fig. 4.

The number of domains in the test set (see Materials and Methods) that are correctly assigned by the MBA method but cannot be identified by PSI-BLAST as a function of CFM (solid squares) and CMK (open squares). Compare those to 726 domains that can be identified in a PSI-BLAST search.
The simplest way of combining keyword- and sequence-based assignments can be realized by adding the motif- and sequence-derived scores according to the formula
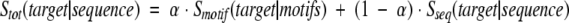 |
(4) |
where Smotif and Sseq are motif- and sequence-based components of the total score Stot and α is an empirically adjustable weight. As shown in Figure 5a ▶ (solid circles), a mixed scoring function that uses Smotif at a CMK = 0.25 cutoff performs, at low values of α, significantly better than does each component alone. Additional improvement of performance is possible by using both CMK and CFM cutoffs. As shown in Figure 5a ▶ (open circles), the accuracy of the combined method can be as high as 95% at 31% coverage (α = 0.075,CMK = 0.25, CFM = 4.0).
Fig. 5.

(a) The performance of the MBA method using the combined motif and sequence scoring (equation 4). The accuracy versus coverage (see Materials and Methods) curve (solid circles) is parametrized by 0 < α < 1 for CFM = 0.25. Additional gain in accuracy can be obtained by also applying annotation filtering 0CMK < 6 (open circles). (b) The cumulative performance of PSI-BLAST and MBA methods using the combined motif and sequence scoring (equation 4). The accuracy versus coverage curve (solid circles) is parametrized by α (equation 4) for CFM = 0.25. Additional gain in accuracy can be obtained by also applying annotation filtering 0CMK < 6 (open circles). α parameter changes are between 0 and 1 along the closed symbols lines. CMK changes along open symbols lines for a fixed value of α.
Interestingly enough, the performance of the Stot score is better than that of Sseq alone even when α = 0. In this case, the presence of motifs affects the fold assignment only through CMK and CFM cutoffs but does not modify the initial, sequence score-based ranking of the targets. Note that such a scoring scheme is analogous to the use of the occurrence of highly conserved sequence motifs during manual analysis of protein sequence. Thus, the improvement observed for α = 0 is consistent with the usefulness of this common, manual approach.
Despite the partial reliance of the combined score on the sequence information, the set of predictions based on Stot is still different than the results returned by PSI-BLAST. Thus, combining the two methods results in an additional increase of the performance shown in Figure 5b ▶. Here the prediction was generated by first running PSI-BLAST and accepting hits at a significance level of p = 1 × 10−3. When PSI-BLAST returned no significant hits, a combined sequence–motif assignment was generated. It is apparent that, at the accuracy level of 95%, the coverage of the combined method is more than five times higher than for SDP and about 40% higher than for PSI-BLAST (50% vs. 35% total coverage).
Discussion
The use of sequence motifs to analyze protein structure and function is one of the most common ways of analyzing novel sequences. Very often the first step in sequence analysis is a similarity search against databases of known sequences such as SwissProt, PIR or GeneBank, followed by identification of the conserved regions that, presumably, are functionally or structurally important. The significance of the resulting motifs is then tested experimentally and by a literature search and comparison against libraries of known sequence motifs, such as Prosite, Blocks, Prints, and others. Until recently, such analyses were performed on a case-to-case basis, relying, to a large extent, on the knowledge of human experts. Such an approach, although feasible on a scale of a few sequences, does not scale well to the number of sequences produced by the genome-wide projects.
The automated motif-based fold assignment method presented here is based on two observations. First, the observation that the functional sites are more often conserved than the rest of the sequence establishes a traceable correlation between folds and sequence motifs, even in cases where automatic detection of the sequence–sequence homology is not reliable. Second, the observation that functional annotation can be used to identify "meaningful" motifs allows one to filter them out from random occurrences inevitable for information-poor, short motifs. The use of the two independent criteria—annotation and motif occurrence—to obtain the motif-based score bypasses the problem frequently encountered when dealing with remote homologs: the decrease of coverage that accompanies eliminating false positives by raising the significance cutoff. The scoring scheme presented here uses the correlation between two partially independent sources of information and thus is less compromised by uncorrelated noise in either of them.
The contribution of functional annotation to fold assignment is helpful for a number of reasons. The most significant is that motifs shared by only a few folds or present in only a subset of folds can be identified by the virtue of the annotation–motif correlation. This allows for a less stringent motif–fold cutoff, CFM, leading to increased coverage of the method without sacrificing accuracy. Another advantage is the possibility of using motifs present in the sequences closely related to the probe, such as identified through BLAST searches. Those motifs, although by definition not completely conserved, can still provide information about the possible functional sites of the fold to be identified. This information can be further validated using the annotation–motif correlation. Initial results indicate that the performance of the modified method is at least comparable to the use of motifs present only in the original probe sequence (data not shown).
In the benchmark adopted here, we attempted to eliminate any effects of prior knowledge of the probe structure on the results. We assumed that no annotation of the probe sequence is available, either directly or through a simple BLAST search of annotated sequence databases or through other forms of function prediction (Marcotte et al. 1999; Pellegrini et al. 1999; Marcotte 2000) or data-mining techniques (Andrade et al. 1999). However, the final version of the algorithm can easily accommodate and benefit from additional annotation of probe sequences obtained experimentally, through a literature search (see MacCallum et al. 2000) or as the result of functional predictions. In the latter case, the predictions are often inferred in a sequence-independent manner (Marcotte 2000). Annotation obtained in this way is often independent from sequence- and experiment-based information that is used by the current version of the algorithm, and would therefore be expected to enhance the signal.
It should be pointed out that, our method, although using functional annotation, does not rely directly on the annotation transfer between homologous proteins, but, rather, detects correlations between annotation and sequence motifs. Thus, it is not limited by a low level of function conservation that has been reported recently (Devos and Valencia 2000; Wilson et al. 2000), and, at the same time, is relatively insensitive to random annotation errors that are not correlated with the motif presence. Such an approach is in contrast to the recently introduced Fuzzy Functional Forms of Fetrow et al (Fetrow and Skolnick 1998) and SiteMatch method of Zhang et al. (1999), both of which are based on recognition of conserved spatial or sequence motifs to identify a protein's function after initial fold assignment. It can be expected that these methods, although efficient at intermediate and high homology levels, might suffer from the alignment errors often encountered in low homology alignments (Domingues et al. 2000b).
In short, the MBA method combines, in a completely automatic way, information provided by occurrences of sequence motifs with functional annotation. The only other method that uses functional information is SAWTED (MacCallum et al. 2000), which relies exclusively on the annotation of the probe sequence. Probe annotations are, obviously, more direct and accurate sources of functional information about the probe sequence than is the annotation of the target domain. However, it is difficult to ensure that the knowledge about the probe's structure does not influence the annotation of the target domains. These factors, together with differences in the benchmarking methodology, make a direct, quantitative comparison of the methods difficult.
Currently, the MBA method is limited by a small number (∼1300) of the motifs defined in the Prosite database. In addition, at a 95% level of accuracy (i.e., CMK = 0.25, CFM = 4.0), only ∼25% of those can contribute to Stot, as correlated, at a high enough level (i.e., SFMCFM) with at least one domain in the fold library. This limitation could be overcome by using large, automatically generated motif libraries, such as those created by EMOTIF (Nevill-Manning et al. 1998) or TEIRESIAS (Rigoutsos et al. 1999), because it should be expected that, the larger the size of the library, the more of the structural and functional features of a fold will be captured. However, the specificity of the motif libraries, in general, decreases with their size, and thus identification of false positives becomes a problem. We hope that a combination of the filtering criteria used in this work will maintain the high accuracy of the method as its coverage is increased.
Materials and methods
Databases
The 8/20/99 version of the NCBI NR BLAST database (405,485 nonredundant sequences) was downloaded and searched for the presence of structural motifs defined in the 1.6 release of the Prosite database (Hofmann et al. 1999). Structures present in the May 1999 release of PDB Select (Hobohm and Sander 1994) were split into domains according to the CATH database (version 1.7 beta; Orengo et al. 1999) and grouped into folds based on the Class, Architecture, and Topology parameters assigned to each structural domain. SwissProt (release 39; Bairoch and Apweiler 2000) keyword annotation was used as the sole source of functional data.
Fold library
The set of 3076 domains representing 522 distinct folds (as defined by CATH classes with a unique Class:Architecture:Topology identifier; 246 folds were represented by more than one structure) was created as a subset of all CATH domains present in both PDB Select and NR BLAST databases. Representatives of the discontinuous folds and all transmembrane domains were discarded.
Sequence/structure compatibility score
The sequence–secondary structure profile method (SDP) was used for the initial sequence/structure prediction as described earlier (Fischer and Eisenberg 1996). Briefly, a Gonnet substitution matrix (Gonnet et al. 1992) was used as a sequence-dependent component of the scoring function, whereas the secondary structure-dependent component was based on a secondary structure substitution matrix calculated as described by Rice and Eisenberg (1997). GLOLOC modification of the Smith-Waterman algorithm (Fischer and Eisenberg 1996) was used to generate sequence-profile alignments using 4.5 and 0.5 for gap opening and extension penalties, respectively.
PSI-BLAST score
NCBI implementation of the PSI-BLAST algorithm was used to assign folds following the methodology of Muller et al. (1999). Briefly, the NR BLAST database was combined with all the contiguous domains from the CATH database. After removal of the low-complexity regions (Wootton and Federhen 1996) up to 20 iterations of PSI-BLAST were performed to obtain a list of domain hits ranked by e-value. Only hits with an e-value <1 × 10−3 were accepted. Drift of the PSI-BLAST searches was avoided by adjusting the value of the h parameter as described by Muller and coworkers (1999).
Motif-based score
The motif-based score was calculated as
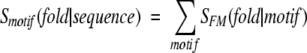 |
(5) |
where SMF was calculated by equation 1 and summation is performed over all motifs found in the evaluated sequence and fulfilling one of the two criteria
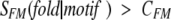 |
(6) |
or:
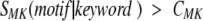 |
(7) |
where CFM and CMK are adjustable parameters and SMK is calculated according to equation 2.
In practice, because of the small size of the motif library, the sum (5) is typically reduced to one component.
Performance benchmark
To evaluate the performance of the fold assignment methods, we scored all of the sequences containing the domains in the fold library (probes) scored against all of the library domains (targets). The ranked prediction list was screened to remove self-hits and domains considered too similar to the structural domains identified in the probe sequence. As a similarity criterion, the relative positions within the CATH hierarchy of the target domain and the domains within the probe sequence were used. Namely, the CATH numerical identifier of the target domain had to differ at, at least, one of the top five levels of the CATH hierarchy (i.e., Class, Architecture, Topology, Homology, and Superfamily) from the probe domain to be taken into consideration. It was also required that, while constructing the SFM table, only domains fulfilling the above criterion were used.
The prescreened list of the hits was used to generate the best prediction by selecting a set of the top-ranked targets covering the entire length of the probe but overlapping <25% of their length. A prediction was considered to be correct (true positive) when the target domain had at least a two-thirds sequence overlap with the probe domain and identical Class, Architecture, and Topology identifiers. Any difference in the identifiers at such a level of overlap was considered a misprediction (false positive), whereas predictions overlapping over less than two-thirds of the length were considered neutral and not taken into account.
Performance of prediction methods is presented in the form of accuracy versus coverage curves, in which accuracy is the ratio of the number of true positives to the total number of predictions and coverage is reported relative to the number of domains (2381) in the probe library with at least one remote homolog (see earlier) present in the target library.
The initial tests at different levels of similarity within CATH have shown that the enforcing difference at the top four levels renders the benchmark too difficult for the current prediction methods (2% PSI-BLAST coverage), whereas releasing the stringency by enforcing the difference at as much as the top six levels resulted in >65% of probe domains assignable with PSI-BLAST.
Acknowledgments
We thank R. Grothe and I. Xenarios for discussions and DOE and NIH for support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
3D, three-dimensional
HMM, hidden Markov model
SDP, sequence-derived properties
MBA, motif-based fold assignment
TIM, triose phosphate isomerase
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.14401.
References
- Altschul, S.F., Madden, T.L., Schaeffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade, M., Casari, G., de Daruvar, A., Sander, C., Schneider, R., Tamames, J., Valencia, A., and Ouzounis, C. 1997. Sequence analysis of the Methanococcus jannaschii genome and the prediction of protein function. Comput. Appl. Biosci. 13 481–483. [DOI] [PubMed] [Google Scholar]
- Andrade, M.A., Brown, N.P., Leroy, C., Hoersch, S., de Daruvar, A., Reich, C., Franchini, A., Tamames, J., Valencia, A., Ouzounis, C., and Sander, C. 1999. Automated genome sequence analysis and annotation. Bioinformatics 15 391–412. [DOI] [PubMed] [Google Scholar]
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxevanis, A.D. 2000. The molecular biology database collection: An online compilation of relevant database resources. Nucleic Acids Res. 28 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienkowska J.R., Yu, L., Zarakhovich, S., Rogers Jr., R.G., and Smith, T.F. 2000. Protein fold recognition by total alignment probability. Proteins 40 451–462. [DOI] [PubMed] [Google Scholar]
- Bork, P and Koonin, E.V. 1996. Protein sequence motifs. Curr. Opin. Struct. Biol. 6 366–376. [DOI] [PubMed] [Google Scholar]
- Bowie, J.U., Luthy, R., and Eisenberg, D. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253 164–170. [DOI] [PubMed] [Google Scholar]
- Devos, D. and Valencia, A. 2000. Practical limits of function prediction. Proteins 41 98–107. [PubMed] [Google Scholar]
- Domingues, F.S., Koppensteiner, W.A., and Sippl, M.J. 2000a. The role of protein structure in genomics. FEBS Lett. 476 98–102. [DOI] [PubMed] [Google Scholar]
- Domingues, F.S., Lackner, P., Andreeva, A., and Sippl, M.J. 2000b. Structure-based evaluation of sequence comparison and fold recognition alignment accuracy. J. Mol. Biol. 297 1003–1013. [DOI] [PubMed] [Google Scholar]
- Fetrow, J.S. and Skolnick, J. 1998. Method for prediction of protein function from sequence using the sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J. Mol. Biol. 281 949–968. [DOI] [PubMed] [Google Scholar]
- Fischer, D. and Eisenberg, D. 1996. Protein fold recognition using sequence-derived predictions. Protein Sci. 5 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnet, G.H., Cohen, M.A., and Benner, S.A. 1992. Exhaustive matching of the entire protein sequence database. Science 256 1443–1445. [DOI] [PubMed] [Google Scholar]
- Gribskov, M., McLachlan, A.D., and Eisenberg, D. 1987. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. 84 4355–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi, H. and Gerstein, M. 1999. The relationship between protein structure and function: A comprehensive survey with application to the yeast genome. J. Mol. Biol. 288 147–164. [DOI] [PubMed] [Google Scholar]
- Hobohm, U. and Sander, C. 1994. Enlarged representative set of protein structures. Protein Sci. 3 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, K., Bucher, P., Falquet, L., and Bairoch, A. 1999. The PROSITE database, its status in 1999. Nucleic Acids Res. 27 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.T. 1999. GenTHREADER: An efficient and reliable protein fold recognition method for genomic sequences. J. Mol. Biol. 287 797–815. [DOI] [PubMed] [Google Scholar]
- Jones, D.T., Taylor, W.R., and Thornton, J.M. 1992. A new approach to protein fold recognition. Nature 358 86–89. [DOI] [PubMed] [Google Scholar]
- Jones, D.T., Tress, M., Bryson, K., and Hadley, C. 1999. Successful recognition of protein folds using threading methods biased by sequence similarity and predicted secondary structure. Proteins Suppl 3: 104–111. [DOI] [PubMed]
- Karplus, K., Barrett, C., and Hughey, R. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics 14 846–856. [DOI] [PubMed] [Google Scholar]
- Kasuya, A. and Thornton, J.M. 1999. Three-dimensional structure analysis of PROSITE patterns. J. Mol. Biol. 286 1673–1691. [DOI] [PubMed] [Google Scholar]
- MacCallum, R.M., Kelley, L.A., and Sternberg, M.J. 2000. SAWTED: Structure assignment with text description-enhanced detection of remote homologues with automated SWISS-PROT annotation comparisons. Bioinformatics 16 125–129. [DOI] [PubMed] [Google Scholar]
- Marcotte, E.M. 2000. Computational genetics: finding protein function by nonhomology methods. Curr. Opin. Struct. Biol. 10 359–365. [DOI] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Ng, H.L., Rice, D.W., Yeates, T.O., and Eisenberg, D. 1999. Detecting protein function and protein–protein interactions from genome sequences. Science 285 751–753. [DOI] [PubMed] [Google Scholar]
- Muller, A., MacCallum, R.M., and Sternberg, M.J.E. 1999. Benchmarking PSI-BLAST in genome annotation. J. Mol. Biol. 293 1257–1271. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G. and Bateman, A. 1997. Distant homology recognition using structural classification of proteins. Proteins Suppl. 1: 105–112. [DOI] [PubMed]
- Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48 443–453. [DOI] [PubMed] [Google Scholar]
- Nevill-Manning, C.G., Wu, T.D., and Brutlag, D.L. 1998. Highly specific protein sequence motifs for genome analysis. Proc. Natl. Acad. Sci. 95 5865–5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo, C.A., Jones, D.T., and Thornton, J.M. 1994. Protein superfamilies and domain superfolds. Nature 372 631–634. [DOI] [PubMed] [Google Scholar]
- Orengo, C.A., Pearl, F.M.G., Bray, J.E., Todd, A.E., Martin, A.C., Lo Conte, L., and Thornton, J.M. 1999. The CATH database provides insights into protein structure/function relationships. Nucleic Acids Res. 27 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko, A.R., Marchler-Bauer, A., and Bryant, S.H. 2000. Combination of threading potentials and sequence profiles improves fold recognition. J. Mol. Biol. 296 1319–1331. [DOI] [PubMed] [Google Scholar]
- Pellegrini, M., Marcotte, E.M., Thompson, M.J., Eisenberg, D., and Yeates, T.O. 1999. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. 96 4285–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, D.W. and Eisenberg, D. 1997. A 3D-1D substitution matrix for protein fold recognition that includes predicted secondary structure of the sequence. J. Mol. Biol. 267 1026–1038. [DOI] [PubMed] [Google Scholar]
- Rigoutsos, I., Floratos, A., Ouzounis, C., Gao, Y., and Parida, L. 1999. Dictionary building via unsupervised hierarchical motif discovery in the sequence space of natural proteins. Proteins 37 264–277. [DOI] [PubMed] [Google Scholar]
- Russell, R.B., Copley, R.R., and Barton, G.J. 1996. Protein fold recognition by mapping predicted secondary structures. J. Mol. Biol. 259 349–365. [DOI] [PubMed] [Google Scholar]
- Skolnick, J. and Fetrow, J.S. 2000. From genes to protein structure and function: Novel applications of computational approaches in the genomic era. Trends Biotechnol. 18 34–39. [DOI] [PubMed] [Google Scholar]
- Skolnick, J., Fetrow, J.S., and Kolinski, A. 2000. Structural genomics and its importance for gene function analysis. Nat. Biotechnol. 18 283–287. [DOI] [PubMed] [Google Scholar]
- Smith, T.F. 1999. The art of matchmaking: Sequence alignment methods and their structural implications. Structure with Folding & Design 7 R7–R12. [DOI] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147 195–197. [DOI] [PubMed] [Google Scholar]
- Wilson, C.A., Kreychman, J., and Gerstein, M. 2000. Assessing annotation transfer for genomics: Quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J. Mol. Biol. 297 233–249. [DOI] [PubMed] [Google Scholar]
- Wootton, J.C. and Federhen, S. 1996. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 266 554–571. [DOI] [PubMed] [Google Scholar]
- Zhang, B., Rychlewski, L., Pawlowski, K., Fetrow, J.S., Skolnick, J., and Godzik, A. 1999. From fold predictions to function predictions: Automation of functional site conservation analysis for functional genome predictions. Protein Sci. 8 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]