

Abstract
Two novel crystal structures of Zea mays protein kinase CK2α catalytic subunit, one in complex with the specific inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) and another in the apo-form, were solved at 2.2 Å resolution. These structures were compared with those of the enzyme in presence of ATP and GTP (the natural cosubstrates) and the inhibitor emodin. Interaction of TBB with the active site of CK2α is mainly due to van der Waals contacts, with the ligand fitting almost perfectly the cavity. One nitrogen of the five-membered ring interacts with two charged residues, Glu 81 and Lys 68, in the depth of the cavity, through two water molecules. These are buried in the active site and are also generally found in the structures of CK2α enzyme analyzed so far, with the exception of the complex with emodin. In the N-terminal lobe, the position of helix αC is particularly well preserved in all the structures examined; the Gly-rich loop is displaced from the intermediate position it has in the apo-form and in the presence of the natural cosubstrates (ATP/GTP) to either an upper (with TBB) or a lower position (with emodin). The selectivity of TBB for CK2 appears to be mainly dictated by the reduced size of the active site which in most other protein kinases is too large for making stable interactions with this inhibitor.
Keywords: CK2, casein kinase 2, protein kinase, apo-CK2, emodin, TBB, inhibitors, complexes
Almost 1000 protein kinases were estimated to be encoded by the human genome (Venter et al. 2001), committed to the phosphorylation of about one third of human proteins (Hunter 1994). Considering that many proteins are phosphorylated by more than one protein kinase, it is reasonable to assume that, on the average, each protein kinase impinges on several dozen target proteins. A major challenge in the field is therefore to idey the physiological substrates of each protein kinase. A powerful tool toward this aim would be the availability of highly specific inhibitors affecting just the protein kinase of interest. Because altered functions of individual protein kinases underlie numerous pathological conditions, with special reference to those caused by uncontrolled proliferation (more than half proto-oncogenes encode for protein kinases), these inhibitors may also give rise to new therapeutic agents applicable in different disease indications (Cohen 1999; Garcia Echeverria et al. 2000).
All protein kinases catalyze the same type of reaction and therefore their ATP binding site is highly conserved (Taylor and Radzio Andzelm 1994). Though, a number of variables and sometimes quite unique features surrounding this site in the diverse kinases (Traxler and Furet 1999) give rise to specific pockets and/or docking sites for potential competitive inhibitors. Some of these ATP site-directed inhibitors are currently in the early phase of clinical trials (le Coutre et al. 1999; Senderowicz et al. 1998). A breakthrough toward the rational design of these inhibitors has been provided by the solution of an increasing number of structures of protein kinases in complex with compounds whose binding is competitive with respect to ATP (Battistutta et al. 2000; De Azevedo et al. 1997; Lamers et al. 1999; Schindler et al. 2000).
Protein kinase CK2, previously misnamed "casein kinase-2" for its ability to phosphorylate the aicial substrate casein in vitro, is probably the most pleiotropic member of its family, with >200 substrates currently known (Pinna and Meggio 1997). Many of these are proteins implicated in a wide variety of cellular functions, with special reference to signal transduction, gene expression, and protein synthesis (Allende and Allende 1995; Pinna 1990); these substrates share unique phosphoacceptor sites specified by clusters of acidic residues, located downstream of the amino acids affected by CK2. Another feature of CK2 is its lack of any general mechanism of tight regulation, resulting in high constitutive activity. This, in turn, is suspected to underlie the oncogenic potential of CK2, as has been disclosed by several experimental models (Kelliher et al. 1996; Li et al. 1999; Orlandini et al. 1998; Seldin and Leder 1995), and its possible exploitation by viruses to perform the phosphorylation of proteins essential to their life cycle (for review, see Guerra and Issinger 1999). This includes CK2 among those kinases that are enticing targets for new antineoplastic and antiviral agents.
The CK2 holoenzyme is a tetramer composed by two catalytic (α and/or α`) and two regulatory β-subunits. Whereas association with the β-subunit deeply alters specificity toward phosphoacceptor protein substrates (Marin et al. 2000), the kinetic constants of the catalytic subunits with respect to the cosubstrate ATP are only marginally affected by association with the β-subunit. The crystal structure of maize CK2α subunit (>70% identical to its human homolog) has been solved either in complex with ATP and GTP (Niefind et al. 1998, 1999) or the competitive inhibitor emodin (Battistutta et al. 2000). Specificity of emodin however is rather broad, as it also inhibits a number of other kinases, with special references to receptor tyrosine kinases (Jayasuriya et al. 1992; Zhang et al. 1998). In contrast, 4,5,6,7-tetrabromo-benzotriazole (TBB or tetrabromo-2-azabenzimidazole), originally developed to discriminate between CK2 and CK1 (Szyszka et al. 1995), has been recently shown to display a striking selectivity toward just CK2 among 33 protein kinases tested, either Ser/Thr or Tyr specific (Sarno et al. 2001). This makes TBB the first-choice compound for disclosing the structural features underlying susceptibility of CK2 to specific inhibitors. Toward this aim we have now solved the structure of the complex between CK2α and TBB and compared it with that of unoccupied apo-CK2α and with complexes with ATP and emodin.
Results and Discussion
Apo-CK2α
As a whole, the structure model of CK2α without any ligand in the active site is similar to that crystallized in the presence of ATP or GTP (Niefind et al. 1999). Some local differences can be observed in the active site region, where the space otherwise occupied by the ligand is partially filled by the side chain of Met 163 and some solvent molecules. In the apo form, the position of the Gly-rich loop, residues 42–50, is similar to that in the ATP/GTP-bound molecules, whereas in the presence of inhibitors, either emodin or TBB, it moves toward or away the N-terminal lobe, respectively (Fig. 1 ▶). The binding of the natural cosubstrates does not seem to cause a significant conformational change in the proximity of the catalytic site, so that the protein does not have to modify itself to accommodate the nucleotides molecules.
Fig. 1.

Changes in loop 42–50 position induced by different ligands. Superimposition of Gly-rich loop (residues 42–50) and of helix αC of CK2α in presence of cosubstrate ATP (red), in the Apo-form (yellow), and with the inhibitors TBB (green) and emodin (cyan). The main difference is found in loop 42–50, while helix αC is perfectly superimposed in all four structures.
CK2α–TBB complex
Crystals of the complex between Zea mays CK2α catalytic subunit and the inhibitor TBB are triclinic, space group P1. They contain two molecules in the asymmetric unit, related by a noncrystallographic twofold axis. In contrast, all of the other structures of the same enzyme solved to date belong to the monoclinic space group C2 and are isomorphous. The complex CK2α–TBB was obtained by soaking the protein crystals, grown in conditions that usually gave the monoclinic crystals, with a solution containing the inhibitor. It is not possible to establish whether the crystals we have measured were already triclinic before incubation with TBB or whether the crystal form has changed as a consequence of the long soaking, resulting in the modification of the crystal packing induced by the inhibitor. The latter hypothesis can be feasible because in the triclinic cell there are two molecules related by a twofold noncrystallographic axis. The transformation from space group C2 to P1 implies a relative reorientation of the two molecules that in the monoclinic form are related by the crystallographic rotation axis, with a consequent rearrangement of the molecular packing. The higher level of flexibility of regions 102–110 and 70–74 (see below) in the TBB complex could be considered one of the factors that favor this rearrangement.
The two molecules in the unit cell are very similar. The bilobal-shaped structure is comparable to that reported previously for CK2α, with a more flexible N terminus domain (the r.m.s. between the two molecules in the asymmetric unit is 1.28 Å) and a structurally more rigid C terminus one (r.m.s. = 0.50 Å). In particular, loop 102–110 and loop 70–74, that precedes helix αC, have high B-factors and a poorly defined electron density in both molecules, which highlight their flexibility.
The superposition of the Cα backbone of different CK2α complexes indicates that the r.m.s. value never exceeds 0.94 Å (Table 1), showing a good correspondence and indicating that the presence of a ligand in the active site does not deeply affect the overall structure. Subtle differences among the apo and the liganded models are nevertheless present. In particular, although the backbones of the C terminus domain are nearly superimposable in all the structures, the N-terminal domain is more flexible and carries the major modifications. The only element of this domain that is structurally conserved is helix αC, which contributes to the recognition of the peptide substrate (Sarno et al. 1996) (Fig. 1 ▶).
Table 1.
Root mean square deviations among Cα of different CK2α structures (Å)
| APO-CK2 on TBB-CK2 | 0.94 |
| ATP-CK2 on TBB-CK2 | 0.76 |
| GTP-CK2 on TBB-CK2 | 0.78 |
| Emo-CK2 on TBB-CK2 | 0.79 |
The electron density of the inhibitor is well defined and it is almost identical in both molecules present in the asymmetric unit (Fig. 2 ▶). It occupies a position in the active site that belongs to the natural cosubstrate purine moiety. The protein–inhibitor interactions are almost completely hydrophobic (involving Val 45, Val 53, Ile 66, Val 95, Phe 113, Val 116, Met 163, Ile 174), in such a way that TBB fits perfectly into the pocket (Table 2 and Fig. 3A ▶). The only polar interaction involves an inhibitor nitrogen (N1) that is hydrogen bonded to a solvent molecule (W1) that in turns interacts with another solvent molecule (W2) that is hydrogen bonded to Oɛ2 of Glu 81 (Fig. 2 ▶). These two water molecules are nearly in the same position in both α subunits with a relatively low B-factors (around 30 Å2), suggesting their relevant role in the formation of electrostatic contacts between ligand and enzyme.
Fig. 2.

Electron density map of TBB molecule. The |2FO−FC| electron density map at 1 sigma contour level is shown around the inhibitor. Most significant residues and the two water molecules W1 and W2 discussed in the text are depicted. Hydrogen bonds and electrostatic contacts are indicated by broken lines. In the box the chemical formula and the atom numbering of TBB are illustrated.
Table 2.
Main interactions distances between residues of CK2α and TBB
| Residue atom | TBB atoma | Distanceb (Å) | |
| Hydrophobic pocket | |||
| Val45 | Cγ2 | BR5 | 3.5 |
| Val53 | Cγ2 | C4 | 3.6 |
| Ile66 | Cγ2 | BR7 | 3.9 |
| Lys68 | Nɛ | N2 | 3.3 |
| Val95 | Cγ2 | BR7 | 4.2 |
| Phel13 | Cδ2 | BR7 | 4.4 |
| Val116 | Cβ | BR6 | 4.1 |
| Met163 | Cɛ | BR5 | 4.0 |
| Ile174 | Cδ1 | C7 | 3.7 |
| Hydrogen bonds | |||
| W1 | N1 | 2.7 | |
a For the numbering of TBB atoms refer to Fig. 2 ▶.
b In the case of the hydrophobic pocket, it is the minimum distance between a carbon atom of residues forming the hydrophobic pocket and TBB.
Fig. 3.
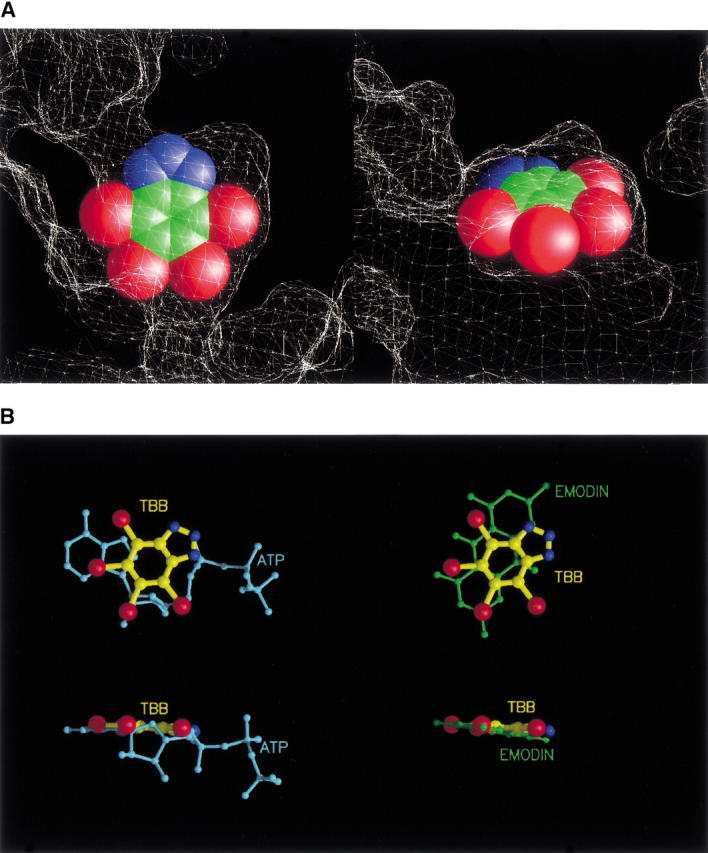
(A) Two clipped views of the active site showing the inhibitor (as cpk model) fitting the cavity are shown. The molecular surface of the protein is represented as a white mesh. Bromine atoms are in red, carbon atoms in green, and nitrogen atoms in blue. (B) Position of TBB with respect to that of ATP (cyan) and emodin (green) in the catalytic site is shown from different points of view. Inhibitor rings lay practically in the same plane of the purine moiety of the natural cosubstrates (bottom).
It is worth noticing that these two water molecules are generally present in all the structures solved to date, with the notable exception of the CK2α–emodin complex (Battistutta et al. 2000), where they are replaced by a portion of emodin itself. Otherwise, these two solvent molecules fill up the active site pocket and strengthen the interactions between the protein and the ligands through hydrogen bonds formation. Two other important water molecules, that allow the "dual-cosubstrate specificity" of CK2 for either ATP or GTP (Wat350 and Wat352 in Niefind et al. 1999), are not present in the CK2α–TBB complex. A superimposition of the three complexes shows that Wat350 should be positioned near Br4 (2.6 Å) and Wat352 between Br5 and Br6 (3.6 Å). The lack of good hydrogen bond acceptors or donors juies the absence of these molecules from the active site of CK2α–|gTββ.
TBB and emodin lay roughly on the same plane of the purine base of the cosubstrates ATP and GTP (Fig. 3B ▶). However, the interactions between the two inhibitors and the protein are mainly hydrophobic, whereas the purine rings of ATP and GTP form some important hydrogen bonds with the polypeptide backbone of CK2α.
Met 163, His 160, and Gly-rich loop
Two residues whose side chain position is critically affected by the presence of either inhibitors or cosubstrate are Met 163, in the catalytic pocket, and His 160, located at the entrance of catalytic site. When the catalytic pocket is occupied, Met 163 side chain is folded with the methyl group pointing down to leave space to the ligand. This invariably happens with ATP, GTP, TBB, and emodin; in contrast, when the cosubstrate site is empty, as in apo-CK2α, the side chain of Met 163 stretches out and the methyl group points into the cavity.
His 160 side chain is also affected by the presence of the ligand: If ATP is present, its triphosphate, protruding outside the active site, keeps the imidazole ring of His 160 turned down, whereas in the CK2α–emodin complex, where the inhibitor is deeply embedded into the cavity, the same ring is turned back and forms an H-bond with the carbonyl oxygen of Arg47. The latter movement also has the effect of blocking emodin inside the active site cavity (Battistutta et al. 2000). With TBB, His 160 side chain has the same orientation as in the presence of emodin, but, in this case, the Gly-rich loop is too far away for any H-bond to be established between His 160 and Arg 47. This highlights once more the flexibility of the Gly-rich loop, which moves from the position it has in the CK2α–ATP complex and in apo-CK2 to either a lower or an upper position, depending on the nature of the ligand (Fig. 1 ▶). In particular, the large size of the bromines of TBB push the loop to an upper position, whereas in the presence of emodin the loop slides down, blocking the entrance to the active site.
TBB selectivity
As reported in a recent study on TBB tested against a panel of 33 different protein kinases (Sarno et al. 2001), this small and relatively simple molecule displays a marked selectivity toward CK2. A comparison of the X-ray structures known for the tested kinases with CK2α reveals that the active site of the latter is invariably smaller in size. This is due to some bulky side chains that in CK2α reduce the space available to cofactors or inhibitors; these are Ile(Val) 66, Met 163, and Ile 174, which in most protein kinases other than CK2 are substituted by less bulky amino acids: Ala versus Ile 66, Leu/Val versus Met 163, Ala/Thr/Leu versus Ile 174. The almost perfect fitting of TBB into the cavity of CK2α active site can explain the selectivity of this molecule. A mutant in which Ile(Val) 66 was substituted with an alanine proved less susceptible to inhibition by TBB, supporting the relevance of the dimension of the cavity for the selectivity of TBB.
Conclusions
TBB is the most specific inhibitor of CK2 known to date and, more in general, one of the most selective inhibitors of protein kinases ever reported (Sarno et al. 2001). The data presented disclose the structural basis for such a remarkable selectivity by showing that TBB is filling a hydrophobic pocket that is present in all protein kinases but whose shape and size in the catalytic subunits of CK2 is almost perfectly tailored for accommodating the molecule of the inhibitor.
This cavity, which is adjacent to and partially overlapping the ATP binding site, is not generated on TBB binding, but it pre-exists as such in the structure of the "empty" CK2α apoenzyme. Its bottom is occupied by two water molecules, found in all the CK2 structures solved to date with the only exception of the structure with emodin where their place is occupied by the inhibitor. These water molecules bridge one of the nitrogen atoms of the triazole ring to Glu 81, stabilizing the structure of the complex. The main interactions between TBB and CK2α however are hydrophobic in nature and in particular they involve the four bromine atoms whose size perfectly fits into the cavity, hampering the release of the inhibitor, once entrapped in it. Indeed, the entrance of TBB into the hydrophobic grove causes minor conformational changes as reflected by the lifting up of the glycine-rich loop from its usual position to an upper one. Consistent with this view, the inhibitory power decreases if the four bromine atoms of TBB are replaced by the smaller chlorine atoms (Szyszka et al. 1995).
It appears therefore that TBB looses most of its inhibitory power with the majority of protein kinases simply because the hydrophobic cavity is generally larger than it is in CK2, failing to detain the TBB molecule, which can freely move inwards and outwards. Several of the CK2 residues forming the hydrophobic pocket where TBB is entrapped are conserved in other kinases as well. Three notable exceptions however are provided by Ile/Val 66, which is invariably an alanine in all protein kinases other than CK2; Ile 174, which is almost always replaced by smaller and less hydrophobic residues (notably Ala, Thr, Gly); and Met 163, whose structural homologs in most kinases have significantly shorter side chains, with predominance of leucine.
The bulkiness of these three unique CK2alpha/alpha` residues appears to be responsible for the reduced size of the hydrophobic cavity, thus accounting for special susceptibility to TBB inhibition. The mutation of just one of these residues, Val 66 (homolog of maize CK2α Ile 66), to alanine in human CK2α caused alone a 10-fold increase in TBB IC50 value (Sarno et al. 2001). On the other hand, it will be possible to enhance the inhibitory efficiency of TBB by modifications designed to generate additional interactions with residues in the active site of CK2α. In this respect, TBB represents a promising lead for the development of hugely selective and more potent inhibitors of CK2.
Materials and methods
Crystals preparation and data collection
The catalytic α subunit of Z. mays CK2α was expressed in Escherichia coli and purified according to a method described previously (Guerra et al. 1998). Crystals were obtained as described elsewhere (Battistutta et al. 2000), without addition of cosubstrates or inhibitors. Single crystals grew in a week at 292°K, using the sitting-drop vapor-diffusion technique.
The complex of CK2α with the inhibitor TBB (CK2α–TBB) was obtained washing the crystals extensively with the precipitant solution and soaking them for 60 d in the same solution saturated with the poorly soluble compound.
In the attempt to obtain a complex with another inhibitor of the antraquinone family, whose solubility in water is particularly low, after an extensive washing of the crystals as described above, the inhibitor was added to crystallization drops in the solid state. Diffraction data on these crystals were collected, but the structure determined revealed that the inhibitor was not bound. This structure can therefore be considered that of the catalytic α subunit of CK2 in absence of any ligand in the active site (apo-CK2α).
Before mounting, crystals were soaked for a few seconds in a cryoprotectant solution consisting of 48% PEG 3400. Because of their size, both crystals were measured at the X-ray diffraction beam-line of ELETTRA synchrotron facility.
For apo-CK2α, a data set was collected at 100°K with a resolution of 2.18 Å and a completeness of 96%. The wavelength used was 1.2 Å and the crystal-to-detector distance was 200 mm. The crystal is isomorphous to the ATP-bound CK2α, space group C2, with cell parameters a = 142.8 Å, b = 58.5 Å, c = 45.1 Å, and β = 102.6°, one molecule in the asymmetric unit, and a Matthews coefficient VM of 2.3 Å3/Da, for a solvent content of 48%.
The crystal of the complex between Z. mays CK2α and TBB was measured using a wavelength of 1 Å at 100°K and a crystal-to-detector distance of 300 mm. The completeness of data set is 89% at a maximum resolution of 2.22 Å. The latter crystal were found to belong to space group P1, with a = 48.3 Å, b = 55.9 Å, c = 60.3 Å, α = 89.8°, β = 102.5°, γ = 99.3°. This corresponds to a VM of 2.0 Å3/Da and a solvent content of 39% with two molecules in the unit cell.
Structure determination and refinement
The structure of the triclinic compound was determined with the molecular replacement method, using the software AMoRe (Navaza 1994) and the structure of the emodin–CK2α complex as a template. The refinement was carried out alternating automated cycles and manual inspection steps, using the CNS software package (Brunger et al. 1998) and O (Jones et al. 1991). Strict noncrystallographic symmetry was imposed on the two molecules in the asymmetric unit in the initial stages of refinement and restrained noncrystallographic symmetry in the final cycles. The presence of the inhibitor was clear from the beginning of the refinement: A flat electron density in the active site, in the zone usually occupied by ATP, was visible both in the |2FO − FC| and |FO − FC| map. During the final cycles of refinement 231 solvent molecules were added. The final model, whose statistics are reported in Table 3, presents an overall R factor of 20.5 (Rfree 26.8) with a good stereochemistry and no residues in disallowed regions according to the program PROCHECK (Laskowski et al. 1993).
Table 3.
Data collection and final model statisticsa
| Data collection | TBB | APO |
| Maximum resolution (Å) | 2.19 (2.31) | 2.10 (2.21) |
| Independent reflections | 27389 (3783) | 20278 (2902) |
| Multiplicity | 1.7 (1.6) | 3.0 (2.3) |
| ≤I/σ> | 13.5 (7.2) | 4.1 (1.9) |
| Rmerge | 0.035 (0.093) | 0.106 (0.301) |
| Completeness (%) | 87.5 (82.8) | 95.6 (94.6) |
| Final model | ||
| Reflection used in refinement | 26659 | 18085 |
| Protein atoms | 5456 (+26 TBB) | 2728 |
| Solvent molecules | 231 | 97 |
| R/Rfree | 20.5/26.8 | 21.7/24.9 |
| R.m.s. on bonds distances (Å) | 0.008 | 0.007 |
| R.m.s. on bond angles (°) | 1.50 | 1.25 |
a Numbers in parentheses refer to the highest resolution bin.
The apo crystal form was refined starting from the ATP-bound model, deprived of the cosubstrate. No electron density in the active site zone was visible throughout all the refinement process. The refinement was carried out as in the previous case. The final model presents an overall R factor of 21.7 (Rfree 24.9), with good statistics (Table 3) and stereochemistry and all residues in allowed regions.
Coordinates
Coordinates have been deposited in the Protein Data Bank (for the CK2α–TBB complex the accession code is 1J91; for Apo-CK2α the accession code is 1JAM).
Acknowledgments
We are grateful to CNR staff at ELETTRA, Trieste, Italy for help during measurements at the diffraction beam-line and to Prof. David Shugar for providing TBB. This work was supported by the Italian National Research Council, Rome, Italy and from grants from E.U. (Biomed-2, BMH4-CT-0047), Armenise-Harvard Foundation, AIRC, Italian Ministry of Health (Project AIDS), Italian MURST (COFIN 2000), and CNR (Target Project on Biotechnology).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.19601.
References
- Allende, J.E. and Allende, C.C. 1995. Protein kinase CK2: An enzyme with multiple substrates and a puzzling regulation. FASEB J. 9 313–323. [DOI] [PubMed] [Google Scholar]
- Battistutta, R., Sarno, S., De Moliner, E., Papinutto, E., Zanotti, G., and Pinna, L.A. 2000. The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J. Biol. Chem. 27529618–29622. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cohen, P. 1999. The development and therapeutic potential of protein kinase inhibitors. Curr. Opin. Chem. Biol. 3 459–465. [DOI] [PubMed] [Google Scholar]
- De Azevedo, W.F., Leclerc, S., Meijer, L., Havlicek, L., Strnad, M., and Kim, S.H. 1997. Inhibition of cyclin-dependent kinases by purine analogues: Crystal structure of human cdk2 complexed with roscovitine. Eur. J. Biochem. 243 518–526. [DOI] [PubMed] [Google Scholar]
- Garcia Echeverria, C., Traxler, P., and Evans, D.B. 2000. ATP site-directed competitive and irreversible inhibitors of protein kinases. Med. Res. Rev. 20 28–57. [DOI] [PubMed] [Google Scholar]
- Guerra, B. and Issinger, O.G. 1999. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 20 391–408. [DOI] [PubMed] [Google Scholar]
- Guerra, B., Niefind, K., Pinna, L.A., Schomburg, D., and Issinger, O.G. 1998. Expression, purification and crystallization of the catalytic subunit of protein kinase CK2 from Zea mays. Acta Crystallogr. D 54 143–145. [DOI] [PubMed] [Google Scholar]
- Hunter, T. 1994. 1001 protein kinases redux towards 2000. Semin. Cell Biol. 5 367–376. [DOI] [PubMed] [Google Scholar]
- Jayasuriya, H., Koonchanok, N.M., Geahlen, R.L., McLaughlin, J.L., and Chang, C.J. 1992. Emodin, a protein tyrosine kinase inhibitor from Polygonum cuspidatum. J. Nat. Prod. 55 696–698. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kelliher, M.A., Seldin, D.C., and Leder, P. 1996. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase II alpha. EMBO J. 15 5160–5166. [PMC free article] [PubMed] [Google Scholar]
- Lamers, M.B., Antson, A.A., Hubbard, R.E., Scott, R.K., and Williams, D.H. 1999. Structure of the protein tyrosine kinase domain of C-terminal Src kinase (CSK) in complex with staurosporine. J. Mol. Biol. 285 713–725. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structure. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- le Coutre, P., Mologni, L., Cleris, L., Marchesi, E., Buchdunger, E., Giardini, R., Formelli, F., and Gambacorti Passerini, C. 1999. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J. Natl. Cancer Inst. 91 163–168. [DOI] [PubMed] [Google Scholar]
- Li, D., Dobrowolska, G., Aicher, L.D., Chen, M., Wright, J.H., Drueckes, P., Dunphy, E.L., Munar, E.S., and Krebs, E.G. 1999. Expression of the casein kinase 2 subunits in Chinese hamster ovary and 3T3 L1 cells provides information on the role of the enzyme in cell proliferation and the cell cycle. J. Biol. Chem. 274 32988–32996. [DOI] [PubMed] [Google Scholar]
- Marin, O., Sarno, S., Boschetti, M., Pagano, M.A., Meggio, F., Ciminale, V., D'Agostino, D.M., and Pinna, L.A. 2000. Unique features of HIV-1 Rev protein phosphorylation by protein kinase CK2 (`casein kinase-2'). FEBS Lett. 481 63–67. [DOI] [PubMed] [Google Scholar]
- Niefind, K., Guerra, B., Pinna, L.A., Issinger, O.G., and Schomburg, D. 1998. Crystal structure of the catalytic subunit of protein kinase CK2 from Zea mays at 2.1 Å resolution. EMBO J. 17 2451–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niefind, K., Putter, M., Guerra, B., Issinger, O.G., and Schomburg, D. 1999. GTP plus water mimic ATP in the active site of protein kinase CK2. Nat. Struct. Biol. 6 1100–1103. [DOI] [PubMed] [Google Scholar]
- Orlandini, M., Semplici, F., Ferruzzi, R., Meggio, F., Pinna, L.A., and Oliviero, S. 1998. Protein kinase CK2alpha` is induced by serum as a delayed early gene and cooperates with Ha-ras in fibroblast transformation. J. Biol. Chem. 273 21291–21297. [DOI] [PubMed] [Google Scholar]
- Pinna, L.A. 1990. Casein kinase 2: An 'eminence grise' in cellular regulation? Biochim. Biophys. Acta 1054 267–284. [DOI] [PubMed] [Google Scholar]
- Pinna, L.A. and Meggio, F. 1997. Protein kinase CK2 ("casein kinase-2") and its implication in cell division and proliferation. Prog. Cell. Cycle. Res. 3 77–97. [DOI] [PubMed] [Google Scholar]
- Sarno, S., Vaglio, P., Meggio, F., Issinger, O.G., and Pinna, L.A. 1996. Protein kinase CK2 mutants defective in substrate recognition. Purification and kinetic analysis. J. Biol. Chem. 271 10595–10601. [DOI] [PubMed] [Google Scholar]
- Sarno, S., Reddy, H., Meggio, F., Ruzzene, M., Davies, S.P., Donella Deana, A., Shugar, D., and Pinna, L.A. 2001. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (`casein kinase-2'). FEBS Lett. 496 44–48. [DOI] [PubMed] [Google Scholar]
- Schindler, T., Bornmann, W., Pellicena, P., Miller, W.T., Clarkson, B., and Kuriyan, J. 2000. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289 1938–1942. [DOI] [PubMed] [Google Scholar]
- Seldin, D.C. and Leder, P. 1995. Casein kinase II alpha transgene-induced murine lymphoma: Relation to theileriosis in cattle. Science 267 894–897. [DOI] [PubMed] [Google Scholar]
- Senderowicz, A.M., Headlee, D., Stinson, S.F., Lush, R.M., Kalil, N., Villalba, L., Hill, K., Steinberg, S.M., Figg, W.D., Tompkins, A., Arbuck, S.G. and Sausville, E.A. 1998. Phase I trial of continuous infusion flavopiridol, a novel cyclin-dependent kinase inhibitor, in patients with refractory neoplasms. J. Clin. Oncol. 16 2986–2999. [DOI] [PubMed] [Google Scholar]
- Szyszka, R., Grankowski, N., Felczak, K., and Shugar, D. 1995. Halogenated benzimidazoles and benzotriazoles as selective inhibitors of protein kinases CKI and CKII from Saccharomyces cerevisiae and other sources. Biochem. Biophys. Res. Commun. 208 418–424. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S. and Radzio Andzelm, E. 1994. Three protein kinase structures define a common m. Structure 2 345–355. [DOI] [PubMed] [Google Scholar]
- Traxler, P. and Furet, P. 1999. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 82 195–206. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291 1304–1351. [DOI] [PubMed] [Google Scholar]
- Zhang, L., Lau, Y.K., Xi, L., Hong, R.L., Kim, D.S., Chen, C.F., Hortobagyi, G.N., Chang, C. and Hung, M.C. 1998. Tyrosine kinase inhibitors, emodin and its derivative repress HER-2/neu-induced cellular transformation and metastasis-associated properties. Oncogene 16 2855–2863. [DOI] [PubMed] [Google Scholar]