

Abstract
The binding of a nitroxide spin-labeled analog of N-acetyllactosamine to galectin-3, a mammalian lectin of 26 kD size, is studied to map the binding sites of this small oligosaccharide on the protein surface. Perturbation of intensities of cross-peaks in the 15N heteronuclear single quantum coherence (HSQC) spectrum of full-length galectin-3 owing to the bound spin label is used qualitatively to idey protein residues proximate to the binding site for N-acetyllactosamine. A protocol for converting intensity measurements to a more quantitative determination of distances between discrete protein amide protons and the bound spin label is then described. This protocol is discussed as part of a drug design strategy in which subsequent perturbation of chemical shifts of distance mapped amide cross-peaks can be used effectively to screen a library of compounds for other ligands that bind to the target protein at distances suitable for chemical linkage to the primary ligand. This approach is novel in that it bypasses the need for structure determination and resonance assignment of the target protein.
Keywords: Nitroxide spin label, galectin-3, drug design, drug screening, distance mapping
Knowing the geometric relationship of various small molecules that bind to protein surfaces can be an important point of reference in the design of effective inhibitors of protein function (Shuker et al. 1996; Hadjuk et al. 1997). Here, we illustrate a method for providing intermolecular distance information based on the use of spin-labeled analogs of known protein ligands to perturb cross-peaks in a 15N–1H heteronuclear single quantum coherence (HSQC) spectrum in a distance-dependent fashion. Labeling cross-peaks in terms of distance from a primary ligand binding site allows use of those cross-peaks in subsequent chemical shift perturbation screens for secondary ligands that bind at appropriate distances for chemical linkage to the primary ligand.
There are precedents in the literature for using a spin-labeled ligand to gain information about protein–ligand interactions (Kosen 1989; Johnson et al. 1999). The most common application employs nitroxide spin labels incorporated in chemically stable molecules such as 2,2,6,6-tetramethyl-1-piperidine-1-oxyl (TEMPO). The route to distance information makes use of the paramagnetic relaxation of NMR resonances caused by these spin-labeled compounds. The relaxation rate enhancement depends on the distance of the nuclei from the unpaired electron, a property that can be used to map distances of nuclei from the spin label. The unpaired electron–nucleus interaction is fairly long-range, unlike the nuclear Overhauser effect (NOE), which is short range in nature (<5 Å). Hence, distances up to 20 Å can be mapped. This general procedure has been used recently to characterize the nature of the interaction between a cellulose-derived ligand and the cellulose binding domains of β-1,4 glucanase CenC from Cellulomonas fimi (Johnson et al. 1999). Also, there is some related work published recently that employs direct interaction between a spin-labeled first ligand and a set of compounds being screened as a second ligand (Jahnke et al. 2000). Our approach differs from the latter method in that it makes use of 15N-labeled protein to obtain distance-dependent data. 1H–15N HSQC spectra are often well-resolved and can be used subsequently to screen a library of compounds in the absence of spin-labeled ligand.
Here, we illustrate the distance mapping procedure with a nitroxide-labeled disaccharide binding to the carbohydrate binding protein galectin-3. Galectin-3 was chosen because its structure and ligand binding activities are well characterized (Seetharamana et al. 1998; Bachhawat-Sikder et al. 2001) and its NMR assignments known (Umemoto and Leffler 2001); this permits validation of the procedure, even if the intended use in the future is for proteins without known structures or NMR assignments. Moreover, galectin-3 is an interesting target for rational drug design.
Galectin-3 is a member of a family of small cytosolic proteins defined by their affinity for β-galactosides and characteristic amino acid sequence ms; more than ten mammalian members of this family have been reported so far (Cooper and Barondes 1999; Leffler 2001). Because they bind typical extracellular carbohydrates and are secreted by non-classical pathways, most attention has been given to their possible extracellular actions such as binding and cross-linking glycoconjugate ligands, possibly forming supramolecular arrays, to modulate cell adhesion and cell signaling (Hughes 1999; Leffler 2001; Sacchettini et al. 2001). Strong evidence indicates roles for galectins, in particular galectin-3, in the regulation of immunity and inflammation and in cancer, although the precise mechanisms of action remain unclear (Liu 2000; Honjo et al. 2001; Leffler 2001; Lowe 2001). While our approach is intended for use in the absence of structure, availability of such detailed information about the galectin-3 system allows us to test distance-based approaches against more conventional structure-based information.
Galectin-3 shows moderate affinity for N-acetyllactosamine (Kd = 0.2 mM; Seetharaman et al. 1998). N-acetyllactosamine (LacNAc) can be turned easily into a spin label analog, N-acetyllactosamine-TEMPO (LacNAc-TEMPO), using TEMPO. From the titration studies of galectin-3 with LacNAC-TEMPO (see Materials and Methods) an affinity of ∼0.1 mM was estimated for the spin-labeled analog, which is quite similar to that for LacNAc. The TEMPO group contains a nitroxide moiety that is paramagnetic because of the presence of an unpaired electron, providing an efficient mechanism for the relaxation of neighboring nuclei via electron–nuclear dipolar coupling.
Using this derivative, we demonstrate here only the first stage in a drug discovery approach, distance mapping of the protein sites using a spin-labeled ligand. Implementation of screening of the second fragment via perturbation of chemical shifts is fairly straightforward (Shuker et al. 1996) and is not illustrated here.
Results
A comparison of the HSQC spectra of galectin-3 with and without LacNAc-TEMPO (Fig. 1 ▶) shows several residues to be affected strongly by the addition of spin label. The cross-peak at 8.8 ppm (1H) and 125.8 ppm (15N) from residue 184 is barely visible at low contour levels in the spectra of the titration in which 30 times excess spin label was added to galectin-3. The cross-peaks of other residues are affected to a lesser extent, indicating that they are farther away from the spin label. Distances of the backbone nitrogen–proton pairs from the affected residues can be estimated by using the intensity measurements for these residues. There are two ways to estimate distances from intensity-based measurements. One is to use estimates of protein reorientational correlation times (τc) and longitudinal relaxation time (T1) measurements to calculate a distance (Johnson et al. 1999). In this case, the distance between the unpaired electron and a given nucleus can be calculated using Equation 1, in which τc is the correlation time for the molecule, ωH is the Larmor frequency for the amide proton in question, T1 is the longitudinal relaxation time, r is the distance of the nucleus from the unpaired electron, and K is a collection of fundamental constants related to spin properties of the system (K = 1/15*S(S+1)γ2g2β2 = 1.23 × 10−32 cm6s−2).
 |
(1) |
Fig. 1.

Comparison of HSQC plots of (A) galectin-3 with no spin label and (B) galectin-3 with 30 times excess spin label. Crosspeaks in the primary binding site are labeled with corresponding assignments. Crosspeaks in the secondary binding site are labeled with asterisks.
Normally, a factor accounting for the fraction of protein with the spin label bound would also be included. In our case, the protein is titrated with spin label to a point of saturation making the factor one. Lower levels of spin label can be used to probe closer distances of approach, once distances measurable at saturation are determined.
However, T1 measurements are time-consuming and thus pose a limitation to approaches for drug design. Therefore, another fairly simple approach based on obtaining distance estimates from transverse relaxation rate enhancements of resonances affected by the spin label is explored. In this approach, simple 1H–15N HSQC spectra are collected that reflect these enhancements as losses in intensities (volumes) of cross-peaks for amide proton–nitrogen pairs. For resonances affected by the spin label, the enhancement in the transverse relaxation rate (1/T2sl-1/T2nsl) is given by,
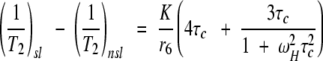 |
(2) |
in which (1/T2)sl corresponds to the transverse relaxation rate in the presence of spin label and (1/T2)nsl corresponds to the transverse relaxation rate in the absence of spin label, τc is the reorientational correlation time for the molecule, ωH is the Larmor frequency for the amide proton in question, r is the distance of the nucleus from the unpaired electron, and K is a collection of constants as described above for equation 1.
Although there are several options for the measurement of transverse relaxation rate enhancement, including those that involve measurement of changes in linewidths (Yu et al. 1994; Johnson et al. 1999) and measurement of peak height ratios (Battiste and Wagner 2000), we opt for a method that uses the following relationship to peak intensities (Johnson et al. 1999) in the presence and absence of spin label.
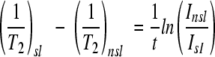 |
(3) |
Here Isl and Insl are the peak intensities (measured as peak volumes) of resonances in the presence and absence of spin label, respectively, and t is the total time during the insensitive nuclei enhanced by polarization transfer (INEPT) and reverse-INEPT periods of the HSQC pulse sequence in which the amide proton magnetization is in the transverse plane and undergoing paramagnetic relaxation. Potential relaxation differences between in-phase and anti-phase proton magnetization are likely to be small and were ignored.
Use of Equation 2 requires knowledge of the correlation time τc for the protein in question. In the absence of any structural information for the target protein, the value of τc can be estimated based solely on knowledge of molecular weight and assumptions regarding internal rigidity and the globular nature of the target protein. The structure of a homologous protein could also be particularly useful in this estimation. Because the electronic lifetime of a nitroxide radical is longer than most protein reorientational correlation times, τc is essentially equal to the reorientational correlation time for the molecule. Using the Stokes equation (Cavanagh et al. 1995), we have estimated a value of τc ∼ 8 nsec for galectin-3 based on its molecular weight of 26 kD.
When measurements of both transverse and longitudinal relaxation rates are available, the value of τc can be calculated directly from the equation:
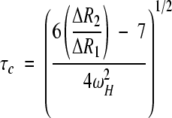 |
(4) |
in which ΔR1 and ΔR2 are the longitudinal and transverse relaxation rate enhancements and ωH is the proton Larmor frequency (Johnson et al. 1999). In our case, values ranging from 5 to 8 nsec are calculated from data on the four most strongly perturbed residues, in reasonable agreement with the molecular-weight-based estimate.
Using the molecular-weight-based estimate of τc and combining Equations 2 and 3, distance estimates for the resolvable resonances affected by the spin label were calculated. Because we know assignments for galectin-3 (Umemoto and Leffler 2001), we can assign these distances to particular residues. This is done in Table 1. Distance estimates were also calculated for other values of τc. Below, we will compare these estimates with numbers based on the structural model and the T1 method (Tables 1,2).
Table 1.
Comparison of distances of residues in the binding site obtained from the X-ray crystal structure model of Galectin-3 (Figure 2) and those calculated with the spin label method (Equation 2)
| Spin label method (Å)b | ||||
| Residue | X-ray model (Å)a | τc = 6 ns | τc = 8 ns | τc = 10 ns |
| 182 | 14.0 | 14.9 | 15.6 | 16.2 |
| 184 | 11.9 | 10.7 | 11.2 | 11.6 |
| 185 | 14.2 | 14.9 | 15.5 | 16.1 |
| 186 | 14.9 | 13.7 | 14.4 | 14.9 |
| 187 | 17.2 | 17.2 | 18.0 | 18.6 |
| 162 | 18.9 | 17.4 | 18.2 | 18.9 |
| 164 | 19.1 | 19.5 | 20.4 | 21.1 |
Distance estimates are tabulated for different values of τc. Distances are compared only for those residues with no resonance overlap and for whom resonance assignments are available.
a) Distances were measured with a precision of ±0.1 Å.
b) Distances were calculated with a precision of ±01. Å (errors in distance calculations were estimated based solely on an error of 2% in measured intensities of cross peaks. Errors involved in estimation of τc or attributable to mobility of TEMPO fragment were not included in the analysis).
Table 2.
Comparison of distances from the X-ray structural model of Galectin-3 (Fig. 2) with distances calculated from T1 relaxation measurements using Equation 1
| Residue | T1 rate enhancement (s−1)a | Distance calc. from T1 rate enhancement (Å)b | X-ray model (Å)b |
| τc = 8 ns | |||
| 182 | 0.061 | 13.4 | 14.0 |
| 184 | 0.193 | 11.1 | 11.9 |
| 185 | 0.058 | 13.6 | 14.2 |
| 186 | 0.045 | 14.1 | 14.9 |
| 187 | 0.018 | 16.5 | 17.2 |
| 162 | 0.012 | 18.1 | 18.9 |
| 164 | 0.008 | 19.2 | 19.1 |
a T1 relaxation rate enhancements were calculated with a precision of ±0.002 s−1.
b Distances were calculated with a precision of ±0.1 Å (errors in distance calculations were estimated based solely on an error of 2% in measured intensities of crosspeaks. Errors involved in estimation of τc or attributable to mobility of TEMPO fragment were not included in the analysis).
Discussion
The X-ray crystal structure of galectin-3 (Seetharaman et al. 1998) shows the primary binding site for LacNAc (Fig. 2 ▶). The amino acid residues in the binding site of galectin-3 are highly conserved in other galectins and are in contact with the LacNAc moeity mainly via hydrogen bonds. Specifically, there are hydrogen bonds involving the C-4 hydroxyl group of the buried galactose moiety; the C-4 hydroxyl group accepts hydrogen bonds from residues His 158 and Arg 162 while donating hydrogen bonds to Asn 160 and a water molecule; the C-6 hydroxyl group from the galactose moiety also forms hydrogen bonds with Glu 184, Asn 174, and another water molecule; and there is van der Waals contact between the other carbon atoms of the galactose and side chains of the aromatic residues in the binding pocket. Unlike the galactose, the N-acetylglucosamine moiety is more solvent-exposed. Only its C-3 hydroxyl group forms hydrogen bonds to Glu 184 and Arg 162.
Fig. 2.

X-ray structure of galectin-3 modeled with LacNAc-TEMPO. The region in dark corresponds to residues affected primarily by the spin label. The distance to the most severely broadened resonance corresponding to residue 184 (shown in green) is indicated by the dashed line.
A peptide bond was used for the linkage of the TEMPO group to the N-acetylglucosamine, not only because it is easy to synthesize, but also because it renders the spin label group less flexible, allowing a more accurate placement of this moiety in a model of LacNAc-TEMPO in the binding site. Because the amide group would extend the TEMPO group away from the protein, the TEMPO group is believed to be largely solvent-exposed and not interacting with any residue in the protein.
Qualitatively, the HSQC resonances perturbed in the presence of LacNAc-TEMPO are consistent with the X-ray model. Residues perturbed strongly by the spin label include residues 160–165 and 182–187. These residues fall within a radius of ∼20 Å from the spin label. The X-ray structure for galectin-3 with LacNAc indicates that residues 162, 165, and 184 are all involved in specific interactions with LacNAc in the binding site (Seetharaman et al. 1998). The most strongly perturbed resonance in the HSQC spectrum belongs to residue 184; it is the residue at the closest distance to the spin label in the X-ray model (Fig. 2 ▶). A more quantitative comparison is presented in Table 1, in which distances measured from the model (N–H pair to N->O bond) are compared with distances calculated using equation 2. Distances were calculated using various values of τc, namely 6, 8, and 10 nsec. Using the 8 nsec value of τc in Equation 2, the distance estimates are in excellent agreement with distances derived from the structural model (RMSD of ±1.5 Å). Even when the values of τc differ by ±2 nsec from the estimated value (i.e., values of 6 and 10 nsec), the distance estimates remain in good agreement, with the RMSD increasing only slightly to ±2 Å. For distances on the order of 20 Å, this corresponds to an error of only ∼10%. Thus, the distance estimates seem to be fairly insensitive to the value of τc used for calculation. This is particularly valuable for structurally uncharacterized and non-globular proteins in which τc values could differ significantly from the estimated value.
Note that the above distances have been calculated based on a very simple procedure, using only intensity measurements in an HSQC spectrum. In Table 2, we compare results using a seemingly more rigorous procedure based on measurement of T1 data (see Results). Again, an estimate of τc = 8 nsec was used in the calculation. The agreement with the X-ray structure model (an RMSD of ±1.0 Å) is only slightly better than that seen in the intensity-based approach.
Distance mapping of protein backbone atoms is but a first step. Once labeled as to distances from the primary ligand site, the HSQC resonances would be used to screen for a second ligand, using the local chemical shift perturbations of resonances belonging to one of the labeled sites. We have not screened for such a second ligand, but have uncovered inadvertently a secondary binding site with some affinity for the TEMPO moiety, either free or attached to LacNAc. During the titration of galectin-3 with LacNAc-TEMPO, resonances from a few residues outside the 20 Å limit predicted from the X-ray model were also observed to suffer line broadening and intensity loss. These included residues 203, 220, 222, 227, 228, and 245. These residues are removed from the primary binding site by 30–35 Å, raising questions about the presence of possible secondary binding sites. When we conducted a second titration using free TEMPO, the resonances from these very same residues (203, 220, 222, 227, 228, and 245) suffered intensity reductions and line broadening. However, the resonances of those in Table 1 were unaffected. Therefore, perturbation of these residues indicates the presence of additional binding sites, perhaps hydrophobic, well removed from the primary binding site found in the crystal structure. In principle, the presence of these binding sites could have been detected by chemical shift perturbations using reduced TEMPO, providing an example of the type of screening that might be done as a second step in a drug discovery protocol. Of greater concern for the present study is that modified primary ligands can have alternate binding sites. Although binding is weak, the affinity for TEMPO could have led to false distance estimates. It will always be prudent to screen with the free TEMPO moiety when using this class of modified primary ligands.
In the normal structure activity relationship (SAR) by NMR approach, the structure of the protein and the additional data from protein–ligand NOEs are sometimes used to determine relative orientation of the fragments, once placement on the protein surface is determined. There are in principle substitutes for these types of data as well. One type of data is residual dipolar couplings collected on ligand spin pairs when the ligand is partially or fully in the bound state. These data can constrain relative orientation of fragments (Prestegard et al. 1999). The combination of distance data and orientational data is quite powerful in that it reduces the number of chemical linkage options that need to be explored for the two fragments.
We have proposed a rapid method for correlating HSQC cross-peaks with distances between N–H pairs and the nitroxide group of a bound spin label as part of a drug design strategy. We have also found an effective spin label for oligosaccharides that can be used in application of this method. The method is useful particularly for proteins lacking structural definition or NMR resonance assignments. Assignment is often the most time-consuming step in protein characterization and often cannot be accomplished for a large protein. Recent advances in NMR (transverse relaxation optimized spectroscopy (TROSY) technique) have allowed collection of HSQC data sets on very large proteins (>100 kD; Pervushin et al. 1997). Without the need for assignments, our method can be extended easily to these large systems.
Materials and methods
Preparation of galectin-3
The protein sample used for the binding studies was full-length galectin-3. This includes a carbohydrate recognition domain and a less structured domain believed to be involved in protein–protein interaction. The expression and purification of galectin-3 was performed using procedures described previously (Massa et al. 1993). Briefly, full-length galectin-3 was expressed in 15N uniformly labeled M9 minimal media using the E. coli strain BL21 (expression vector pet3c). Following standard cell lysis procedures, purification from the supernant solution was done using affinity chromatography on a lactosyl-Sepharose column with elution by lactose. The buffer used in the purification steps was 20 mM sodium phosphate/150 mM NaCl (pH 7.4). Samples were dialyzed versus this buffer (without the lactose) and concentrated to ∼0.3 mM before analysis.
Synthesis of N-acetyllactosamine-TEMPO
Refer to Figure 3 ▶ for the numbered steps in this synthesis. The N-acetyllactosamine starting material [1] was synthesized by the method of Alais and Veyrieres (1990). Synthesis of N-acetyllactosamine-TEMPO was then achieved in two steps. The first step involved the synthesis of the N-acetyllactosylamine (4-O-(β-d-Galactopyranosyl)-2-acetamido-2-deoxy-β-D-glucopyranosylamine) [2] which was synthesized by the general procedure of Likhosherstov et al. (1986), further optimized by methods of Kallin et al. (1989) and Manger et al. (1992). Briefly, 0.200 g N-acetyllactosamine [1] was dissolved in 10 mL water saturated with CO3HNH4 and stirred at room temperature for one week. TLC (CHCl3, CH3OH, and H2O 60:40:10) indicated an almost complete conversion to the glycosylamine [2]. 50 mL water was added and the solvent partly evaporated in vacuum with a bath temperature of 30°–35°C maximum to remove excess ammonium carbonate. This operation was repeated five times. The remaining solution (∼50 mL) was freeze-dried leaving ∼0.25 g of material which was kept at −20°C. The structure of the product was confirmed by NMR.
Fig. 3.

Scheme for the synthesis of LacNAc-TEMPO. (*) Refer to text for numbered compounds.
The second step involved the synthesis of N-acetyllactosamine-TEMPO [4-O-(β-D-Galactopyranosyl)-2-acetamido-1–(1-Oxyl-4-carboxamido-2,2,6,6-tetra-methyl-piperidine)-1,2-deoxy-β-D-glucopyranoside] [5] by condensation of the carboxylated TEMPO derivative [3] with the glycosyl amine of [2] (Fig. 3 ▶). The carboxylated TEMPO derivative, 1-Oxyl-4-carboxyl-2,2,6,6-tetramethylpiperidine [3] was synthesized from 1-oxyl-2,2,6,6-tetramethyl-4-piperidone in two steps, according to the procedure of Rauckman et al. (1971). 1-Oxyl-4-carboxyl-2,2,6,6-tetramethylpiperidine was then activated with 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (Dhbt-OH) in the presence of dicyclohexyl carbodiimide (DCC) and the intermediate [4] condensed with the glycosylamine of N-Acetyllactosamine [2] according a procedure reported by Meinjohanns et al. (1998). The final product was purified by hydrophobic chromatography. Specifically, Dhbt-OH (0.44 mmol, 0.072 g) and DCC (0.44 mmol, 0.090 g) were added to the solution of the starting acid (0.077 g, 0.44 mmol) in dry THF (2 mL). The mixture was stirred at 0°C for 2 h while some precipitate was formed. The insoluble portion was filtered and washed with a small amount of THF. The filtrate was evaporated. More THF (3 mL) was added to the residue and the suspension refiltered. The filtrate was evaporated and quickly dried in vacuum. Dry DMF (1 mL) was added to the solid followed by N,N-Diisopropylethylamine (DIPEA, 0.44mmol, 0.076 mL, 0.056 g) and the crude glycosylamine [2] (0.100 g, 0.293 mmol). The mixture was stirred at room temperature for 3 hours and the progress of the reaction was followed by TLC (CHCl3 CH3OH 80:20 and CHCl3, CH3OH, and H2O 65:35:5). DMF was evaporated in vacuum and water (4 mL) was added. The mixture was left in the refrigerator overnight, and the precipitate filtered and washed with water. The aqueous phase was extracted with CH2Cl2 (2 mL) and the organic layer removed. The operation was repeated 5 times. The remaining aqueous solution was freeze-dried leaving a residue. This residue was applied in water to a column of hydrophobic C-18 silicagel (30 g) conditioned as indicated by the manufacturer (Waters Chromatographic Products) and eluted using a mixture of CH3OH and H2O (10:90, 40 mL). The unreacted amine [2] came out early followed by some colored impurity while the reaction product [5] came out in latter fractions. Evaporation of the appropriate fractions gave 0.119 g of the product (54%).
After reduction of the nitroxide with phenyl hydrazine, the product was characterized by NMR (300 MHz) as follows: Phenyl hydrazine (0.005 mL) was added to the sample (2 mg) in CD3OD. The solvent was evaporated and the sample D2O exchanged. The spectrum was then run in D2O: 5.17 (d, 1H, J = 9.0 Hz, H-1), 4.57 (d, 1H, J = 8.0 Hz, H-1'), 2.85 (tt, 1H, CO-CH), 2.09 (s, 3H, NHAc), 1.65–1.90 (m, 4H, CH2-C(CH3)2), 1.30 (overlapping singlets, 12 H, CH3). Impurity peaks were estimated at less than 5%.
NMR spectroscopy and data analysis
A 0.3 mM protein sample of intact galectin-3 was used in the analysis of the interaction with LacNAc-TEMPO. Additions of LacNAc-TEMPO to the protein sample were made in small amounts from a concentrated stock solution. Any volume changes caused by addition of the spin label were fairly small and did not affect the intensity measurements significantly, as evidenced by the measurement of intensities of cross-peaks not affected by the spin label (>35 Å away based on the available structure of galectin-3). 1H–15N HSQC spectra (Kay et al. 1992) were acquired at 25°C for each of several titration points on a Varian Inova 800 MHz spectrometer collecting 4096 × 256 complex data points in the 1H and the 15N dimensions, respectively. The final ligand-to-protein ratio at each titration point was determined to be 0:1, 0.4:1, 0.8:1, 1:1, 2:1,4:1, 8:1, 18:1, and 30:1. At 30:1, the binding of the ligand to the protein was deemed complete. Intensity measurements at the initial titration point were done on a sample without spin label instead of using reduced spin label to avoid perturbations caused by the presence of ascorbic acid (used as a reducing agent). A separate control experiment (data not shown) involving titration of galectin-3 with just LacNAc showed fairly small intensity changes (<5%) for the residues affected by LacNAc-TEMPO. Therefore, it is reasonable to assume that intensity changes on titration arise purely from paramagnetic perturbations.
The titration of galectin-3 with the TEMPO spin label free of the LacNAc moiety was performed in a similar manner. 1H–15N HSQC spectra (Kay et al. 1992) were acquired at 25°C for each of several titration points on a Varian Inova 800 MHz spectrometer. The final ligand-to-protein ratio at each titration point was determined to be 0:1, 10:1, and 30:1.
T1 relaxation time measurements for backbone amides were performed on a sample without and with the spin label (30:1 ligand-to-protein ratio). A 1H–15N HSQC sequence (Kay et al. 1992) in the form of a non-selective inversion recovery experiment was used to measure the T1 values. The inversion recovery experiments were recorded with delays of 0, 0.05, 0.1, 0.15, 0.2, 0.3, 0.5, 0.7, 1.1, and 1.5 seconds.
Data processing and analysis was performed using the Felix 97.0 software package (Molecular Simulations Inc., San Deigo, CA). Data processing involved convolution difference to minimize baseline distortions near water and multiplication with an exponential function in the directly detected dimension and multiplication with a 90°-shifted squared sinebell function in the indirect dimension. This processing minimizes the distortion of intensities for cross-peaks of different linewidths, making intensity variations primarily a function of T2 decay during the INEPT transfer periods of the HSQC. The resulting matrix after Fourier transformation was then phase-corrected and referenced to yield a two-dimensional spectrum that was used for further analysis. Positions and intensities of cross-peaks (measured as cross-peak volumes) were tabulated using Felix 97.0 software. The relative peak intensities It were fit to the inversion recovery equation It = I0 exp (-t/T1), for different delay times t (Farrow et al. 1994), in which I0 is the peak intensity at time t = 0. T1 rate enhancements were calculated as a difference in the 1/T1 values for the measurements with and without the spin label.
All amide cross-peaks undergoing large intensity changes during titration were ideied. Cross-peaks that showed large changes but were overlapped with other cross-peaks were not included in the analysis, because of difficulties in accurate measurement of their intensities. Also, cross-peaks that showed changes but lacked sequence-specific backbone amide assignments, could not be included in the analysis. The intensities for the ideied cross-peaks were measured and compared with intensities for the same cross-peaks in the dataset in which no spin label was added. Distances from the spin label based on the measured intensities were then calculated for each residue using equations 2 and 3 (see Results).
The LacNAc-TEMPO group was docked into the binding site using the placement of LacNAc in the crystal structure as a template (Fig. 2 ▶; Midasplus software, UCSF). The coordinates for the TEMPO moiety were generated from the bond length, bond angle, and torsion angle values supplied for the TEMPO group in the group library included with the software. The TEMPO group was then modeled onto the LacNAc group in the crystal structure using an amide group as a connector to generate the final LacNAc-TEMPO moiety. Improper modeling of the TEMPO group in LacNAc-TEMPO can lead to errors in distance estimates from the spin label; however, these errors are largely minimized when distances compared are all relatively large. We estimate the error in distance estimates caused by improper modeling to be a maximum of ±1 Å, an insignificant variation compared to the large distances measured. Therefore, it is feasible to use the above-derived model structure of LacNAc-TEMPO for distance mapping from the spin label.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (GM33225 and RR05351).
The publication costs of this article were defrayed in part by payment of page charges.This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.17401.
References
- Alais, J. and Veyrieres, A. 1990. Syntheses of linear tetra-, hexa-, and octa-saccharide fragments of the I-blood group active poly-(N-acetyl-lactosamine) series. Blockwise methods for the synthesis of repititive oligosaccharide sequences. Carbohydr. Res. 207 11–32. [DOI] [PubMed] [Google Scholar]
- Bachhawat-Sikder, K., Thomas, C.J., and Surolia, A. 2001. Thermodynamic analysis of the binding of galacatose and poly-N-acetyllactosamine derivatives of human galectin-3. FEBS Lett. 500 75–79. [DOI] [PubMed] [Google Scholar]
- Battiste, J.L. and Wagner, G. 2000. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 39 5355–5365. [DOI] [PubMed] [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer III, A.G., and Skelton, N.J. 1995. Protein NMR Spectroscopy: Principles and Practice, pp. 17–18. Academic Press, SanDiego, CA.
- Cooper, D.N. and Barondes S.H. 1999. God must love galectins; he made so many of them. Glycobiology 9 979–984. [DOI] [PubMed] [Google Scholar]
- Farrow, N.A., Muhandiram, R., Singer, A.U., Pascal, S.M., Kay, C.M., Gish, G., Shoelsen, S.E., Pawson, T., Foreman-Kay, J.D., and Kay, L.E. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33 5984–6003. [DOI] [PubMed] [Google Scholar]
- Hadjuk, P.J., Sheppard, G., Nettesheim, D.G., Olejniczak, E.T., Shuker, S.B., Meadows, R.P., Steinman, D.H., Carrera, G.M., Marcotte, P.A., Severin, J., Walter, K., Smith, H., Gubbins, E., Simmer, R., Holzman, T.F., Morgan, D.W., Davidsen, S.K., Summers, J.B., and Fesik, S.W. 1997. Discovery of potent nonpeptide inhibitors of stromelysin using SAR by NMR. J. Am. Chem. Soc. 119 5818–5827. [Google Scholar]
- Honjo, Y., Nangia-Makker, P., Inohara, Y., and Raz, A. 2001. Down-regulation of galectin-3 suppresses tumorigenicity of human breast carcinoma cells. Clin. Cancer Res. 7 661–668. [PubMed] [Google Scholar]
- Hughes, R.C. 1999. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim. Biophys. Acta 1473 172–185. [DOI] [PubMed] [Google Scholar]
- Jahnke, W., Perez, L.B., Paris, C.G., Strauss, A., Fendrich, G., and Nalin, C.M. 2000. Second-site NMR screening with a spin-labeled first ligand. J. Am. Chem. Soc. 122 7394–7395. [Google Scholar]
- Johnson, P.E., Brun, E., MacKenzie, L.F., Withers, S.G., and McIntosh, L.P. 1999. The cellulose-binding domains from Cellulomonas fimi β-1,4-Glucanase CenC bind Nitroxide spin-labeled cellooligosaccharides in multiple orientations. J. Mol. Biol. 287 609–625. [DOI] [PubMed] [Google Scholar]
- Kallin, E., Loon, H., Norberg, T., and Elofson, M. 1989. Derivatization procedures for reducing oligosaccharides: Preparation of oligosaccharide glycosylamines and their conversion into oligosaccharide-acrylamide copolymers. J. Carb. Chem. 8 597–611. [Google Scholar]
- Kay, L., Keifer, P., and Saarinen, T. 1992. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114 10663. [Google Scholar]
- Kosen, P.A. 1989. Spin labeling of proteins. Methods Enzymol. 177 86–121. [DOI] [PubMed] [Google Scholar]
- Leffler, H. 2001. Galectins structure and function - A synopsis. Results Probl. Cell Differ. 33 57–83. [DOI] [PubMed] [Google Scholar]
- Likhosherstov, L.M., Novikova, O.S., Derevitskaja, V.A., and Kochetkov, N.K. 1986. A new simple synthesis of amino sugar β-d-glycosylamines. Carb. Res. 146 C1–C5. [Google Scholar]
- Liu, F.T. 2000. Galectins: A new family of regulators of inflammation. Clin. Immunol. 97 79–88. [DOI] [PubMed] [Google Scholar]
- Lowe, J.B. 2001. Glycosylation, immunity, and autoimmunity. Cell 104 809–812. [DOI] [PubMed] [Google Scholar]
- Manger, I.D., Rademacher, T.W., and Dwek, R.A. 1992. 1-N-Glycyl beta-oligosaccharide derivatives as stable intermediates for the formation of glycoconjugate probes. Biochemistry 31 10724–10732. [DOI] [PubMed] [Google Scholar]
- Massa, S.M., Cooper, N.W., Leffler, H., and Barondes, S.H. 1993. L-29, an endogenous Lectin, binds to glycoconjugate ligands with positive cooperativity. Biochemistry 32 260–267. [DOI] [PubMed] [Google Scholar]
- Meinjohanns, E., Meldal, M., Paulsen, H., Dwek, R.A., and Bock, K. 1998. Novel sequential solid-phase synthesis of N-linked glycopeptides from natural sources. J. Chem. Soc. Perkin Trans. 1 549–560. [Google Scholar]
- Pervushin, K., Reik, R., Wider, G., and Wuthrich, K. 1997. Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. 95 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestegard, J.H., Tolman, J.R., Al-Hashimi, H.M., and Andrec, M. 1999. Protein structure and dynamics from field-induced residual dipolar coupling. In Biol. Magn. Reson. (ed. N.R. Krishna and L.J. Berliner), pp. 311–355. Kluwer Academic / Plenum Publishers, New York.
- Rauckman, E.J., Rosen, G.M., and Abou-Donia, M.B. 1971. Synthesis of a useful spin-labeled probe, 1-oxyl-4-carboxyl-2,2,6,6-tetramethyl piperidine. J. Org. Chem. 41 564–565. [Google Scholar]
- Sacchettini, J.C., Baum, L.G., and Brewer C.F. 2001. Multivalent protein–carbohydrate interactions. A new paradigm for supermolecular assembly and signal transduction. Biochemistry 40 3009–3015. [DOI] [PubMed] [Google Scholar]
- Seetharaman, J., Kanigsberg, A., Slaaby, R., Leffler, H., Barondes, S., and Rini, J. 1998. X-ray crystal structure of the human Galectin-3 carbohydrate recognition domain at 2.1 Å resolution. J. Biol. Chem. 273 13047–13052. [DOI] [PubMed] [Google Scholar]
- Shuker, S.B., Hadjuk, P.J., Meadows, R.P., and Fesik, S.W. 1996. Discovering high-affinity ligands for proteins: SAR by NMR. Science 274 1531–1534. [DOI] [PubMed] [Google Scholar]
- Umemoto, K. and Leffler, H. 2001. Assignment of 1H, 15N and 13C resonances of the carbohydrate recognition domain of human galectin-3 J. Biomol. NMR 20 91–92. [DOI] [PubMed] [Google Scholar]
- Yu, L., Meadows, R.P., Wagner, R., and Fesik, S.W. 1994. NMR studies of the FK506 binding protein bound to a spin-labeled ascomycin analog J. Magn. Reson. 104 77–80. [DOI] [PubMed] [Google Scholar]