

Abstract
Glutathione S-transferases (GSTs) are dimeric proteins that play an important role in cellular detoxification. Four GSTs from the mosquito Anopheles dirus species B (Ad), an important malaria vector in South East Asia, are produced by alternate splicing of a single transcription product and were previously shown to have detoxifying activity towards pesticides such as DDT. We have determined the crystal structures for two of these alternatively spliced proteins, AdGST1–3 (complexed with glutathione) and AdGST1–4 (apo form), at 1.75 and 2.45 Å resolution, respectively. These GST isozymes show differences from the related GST from the Australian sheep blowfly Lucilia cuprina; in particular, the presence of a C-terminal helix forming part of the active site. This helix causes the active site of the Anopheles GSTs to be enclosed. The glutathione-binding helix α2 and flanking residues are disordered in the AdGST1–4 (apo) structure, yet ordered in the AdGST1–3 (GSH-bound) structure, suggesting that insect GSTs operate with an induced fit mechanism similar to that found in the plant phi- and human pi-class GSTs. Despite the high overall sequence identities, the active site residues of AdGST1–4 and AdGST1–3 have different conformations.
Keywords: Glutathione S-transferase, induced fit, mosquito, Anopheles dirus species B, pesticide resistance
Glutathione S-transferases (GSTs, EC 2.5.1.18) are phase II detoxifying enzymes that conjugate xenobiotic compounds (e.g., drugs, herbicides, insecticides) with electrophilic centers to glutathione (GSH, γ-glutamyl-cysteinyl-glycine). GSTs also possess functions as diverse as prostaglandin synthesis (Kanaoka et al. 1997) and regulation of intracellular ion channels (Dulhunty et al. 2001). There are two distinct groups of GSTs: the trimeric membrane-associated microsomal enzymes and the soluble, dimeric cytosolic enzymes (Hayes and Pulford 1995). The latter group has been the most extensively studied, with hundreds of sequences known from animals, plants, fungi, and bacteria. Although no archeal GSTs are known, biochemistry involving γ-glutamylcysteine, a GSH precursor, has been described in archea (Sundquist and Fahey 1989). All cytosolic GSTs feature a GSH binding site called the G-site, and an H-site that recognizes the hydrophobic cosubstrate. The H-site shows the greatest variability across GST classes.
Although ten GST classes (with >50% identity within a class) are currently recognized, the known sequences can be organized into at least 25 families (Snyder and Maddison 1997). In mammals, alpha-, pi-, mu-, and theta-class GSTs (Mannervik et al. 1992) and recently kappa- (Pemble et al. 1996) as well as zeta (Board et al. 1997) and omega-class isozymes have been described (Board et al. 2000). Mammalian prostaglandin D-synthases are homologs of the invertebrate sigma-class GSTs (Kanaoka et al. 2000). Insect GSTs were previously assigned to class theta (Pemble and Taylor 1992). It has since been recognized that the insect GSTs are structurally and functionally distinct from the mammalian theta-class GSTs and are now designated as delta class (Board et al. 1997; Ketterman et al. 2001). Regarding the soluble dimeric GSTs, it has been observed that organisms can possess multiple isoforms of various classes of GST (Mannervik et al. 1992). Homo- and heterodimers within classes can be formed. However, no heterodimer combining different classes have been described (Wilce and Parker 1994).
Numerous structures of mammalian GSTs have been described. These include pi-class (Reinemer et al. 1992; Dirr et al. 1994; Garcia-Saez et al. 1994; Oakley et al. 1997, 1998, 1999; Prade et al. 1997; Vega et al. 1998; Ji et al. 1999), mu-class (Ji et al. 1992, 1993, 1994; Raghunathan et al. 1994) and alpha-class (Sinning et al. 1993; Cameron et al. 1995; Xiao et al. 1999). In these families, a conserved tyrosine hydroxyl group is responsible for promoting the formation of a thiolate anion in GSH. It has been demonstrated for alpha-, mu-, and delta-class GSTs that a proton is released by GSH upon binding to the enzyme (Caccuri et al. 1997). An analogous tyrosine residue is present in the squid sigma-class GST (Ji et al. 1995). However, in the human theta-class GST, it is a serine residue that provides the hydroxyl group (Rossjohn et al. 1998a), similar to that found in insect delta- and plant phi-class GSTs (see below).
There has been increasing interest in nonmammalian GSTs such as the bacterial beta-class (Rossjohn et al. 1998b), insect (Wilce et al. 1995), and plant phi-class GSTs (Reinemer et al. 1996; Neuefeind et al. 1997a,b) which play roles in antibiotic resistance, insecticide, and herbicide resistance, respectively. Only one insect GST structure has been determined — the homolog from the Australian sheep blowfly Lucilia cuprina (Lc) (Wilce et al. 1995). These isozymes possess active-site chemistries different from those observed in the aforementioned mammalian GSTs. The insect and plant GSTs possess a serine residue in place of tyrosine in the active site. The serine hydroxyl is thought to function in a fashion analogous to that of the tyrosine hydroxyl group. In the bacterial GST crystal structure, an active-site cysteine was observed to form a mixed disulfide with glutathione (Rossjohn et al. 1998b). This is a mixed disulfide intermediate similar to that found in glutaredoxins (Nordstrand et al. 1999). The recently solved human omega-class GST also forms a similar mixed disulfide with glutathione (Board et al. 2000).
Malaria kills about 2.7 million people per year (Phillips, 2001). Successful preventive measures have focused on control of the mosquito vectors using insecticides such as DDT (dichloro-diphenyl-trichloroethane) (Trigg and Kondrachine 1998). Insecticides such as DDT, diazinon, fenitrothion, and parathion have been recognized as substrates for GSTs (Lamoureux and Rusness 1989). GST isozymes from Anopheles dirus species B (the nomenclature of secondary structure elements in the structures presented here follows that of Wilce et al. [1995]) have been demonstrated to possess dehydrochlorination activity towards the insecticide DDT (Prapanthadara et al. 2000). Insecticide-resistant strains of housefly have been observed to possess elevated levels of GST activity in crude extracts (Clark and Dauterman 1982). In a DDT-resistant strain of Anopheles gambiae, there was an increase in the synthesis of GST isozymes possessing a greater dehydrochlorination activity (Prapanthadara et al. 1993, 1995).
Controlling the mosquito vectors of malaria is of vital importance to world health. By exploring the structure and function of insect GSTs, an understanding of the mechanisms of resistance to important pesticides can be gained. These tertiary structures may also be used as templates for the design of inhibitors that can be used to overcome insecticide resistance. To this end, we have determined the structures of two GST isoforms from Anopheles dirus species B, AdGST1–3 and AdGST1–4 (in insect GST nomenclature, "1" refers to the class and "3" and "4" refer to the isozyme numbers). These GSTs are of further interest because they arise from alternate splicing. Thus, the N-terminal domain of the GSTs derives from the same exon, whereas the C-terminal domain arises from different exons (Pongjaroenkit et al. 2001, Jirajaroenrat et al. 2001). The relationship of gene structure to protein structure is discussed.
Results and Discussion
The AdGST1–3 and AdGST1–4 structures
The final electron density for the AdGST1–3 and AdGST1–4 models is excellent (Fig. 1 ▶). The overall topology of AdGST1–3 and AdGST1–4 is similar to that of LcGST (Fig. 2 ▶). The N-terminal domain (residues 1–78) adopts the canonical thioredoxin fold (with βαβαββα topology) found in all GST structures. There is a cis-proline at position 53 in both AdGST isozymes. This proline, found in all GST structures so far determined, appears to be required for the correct formation of the GSH binding site, which is formed by one face of the N-terminal domain. GSH is observed to bind in the active site of AdGST1–3 (Figs. 1a and 2d ▶ ▶). The active catalytic S9 Oγ is 3.28 Å from the GSH sulfhydryl. This is closer than the equivalent distance in LcGST (3.94 Å). It has been noted that substitution of S9 in LcGST reduces the catalytic turnover to about 0.5% of wild-type enzyme (Caccuri et al. 1997).
Fig. 1.
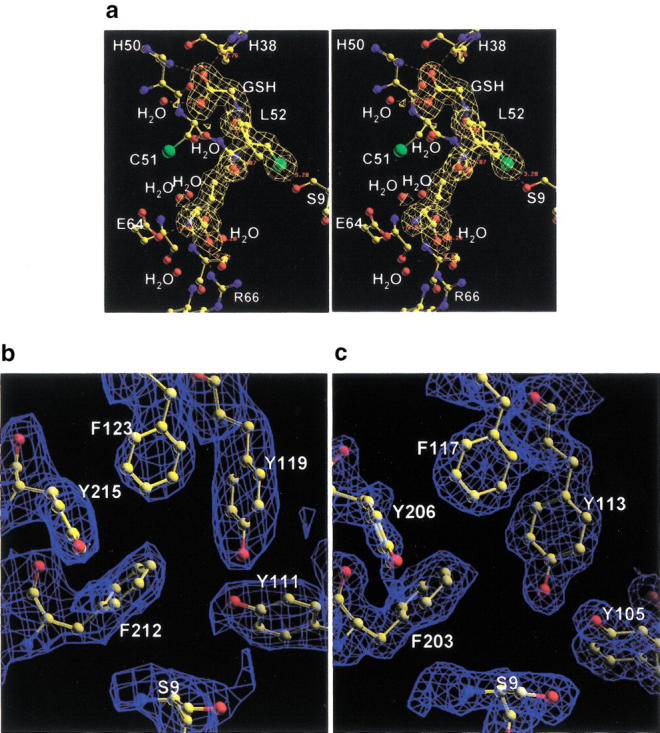
(a) Stereo diagram of electron density of GSH in AdGST1–3 (σa-weighted 2fo-fc map shown in gold at a contour of 1σ) with surrounding G-site residues. Electron density of H-site residues for AdGST1–4 (b) and AdGST1–3 (c) with σa-weighted 2fo-fc maps contoured at 1σ are shown in blue. Ball and stick representation is used for all atoms and bonds.
Fig. 2.
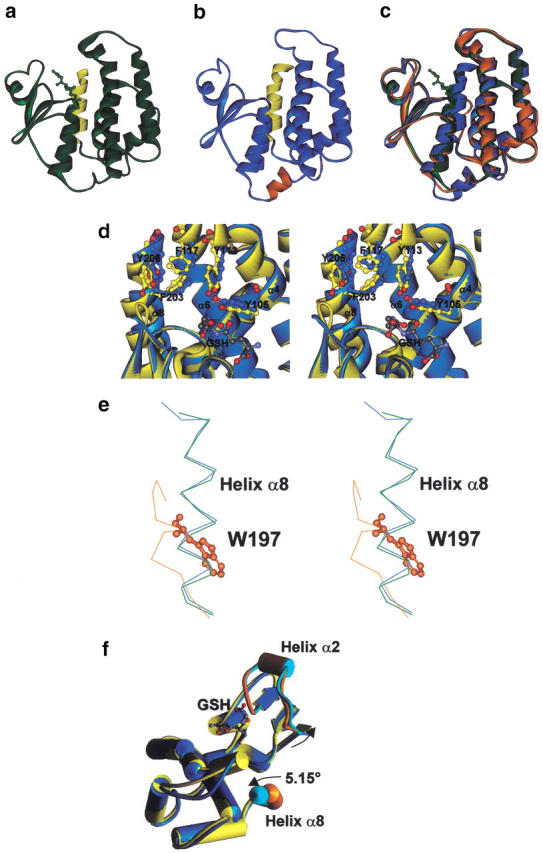
Comparison of AdGST1–3, AdGST1–4, and LcGST structures. Monomers of AdGST1–3 (a) and AdGST1–4 (b) are shown in ribbon form with the five-amino-acid insert in AdGST1–4 between the N- and C-terminal domains colored orange. The C-terminal helices (α8) of both enzymes are colored yellow. (c) Superimposed structures of AdGST1–3 (green), AdGST1–4 (blue), and LcGST monomers (orange). (d) Stereo diagram of the H-sites of AdGST1–3 (yellow) and AdGST1–4 (blue). The fold is represented in ribbon form with putative H-site amino acids shown in ball and stick form. The GSH model is of that found in AdGST1–3. Numbers correspond to residues in AdGST1–3. (e) Stereo diagram of superimposed structures of AdGST1–3 (green), AdGST1–4 (blue), and LcGST (orange) in the vicinity of helix α8. The residue W197 in Lucilia GST overlapping helix α8 in AdGST1–3 and AdGST1–4 is indicated in ball-and-stick form. (f) Comparison of AdGST 1–3 (yellow) versus AdGST1–4 (blue) with helices shown as cylinders. Helix α8 and the region around and including helix α2 is highlighted in orange (for AdGST1–3) and cyan (for AdGST1–4). The shifts in AdGST1–4 relative to AdGST1–3 are indicated.
The C-terminal domains of AdGST1–3 and AdGST1–4 (residue 85–) consist of a bundle of five helices. Four of these align closely with equivalent helices in LcGST (Fig. 2c ▶). Helix α6 is central to these bundles and contains an N-capping box (S/T-X-X-D) and a hydrophobic staple m (Fig. 3 ▶), both of which are highly conserved among GSTs and appear to greatly stabilize the GST fold (Dragani et al. 1997; Stenberg et al. 2000). In AdGST1–4, the first leucine of the staple is substituted for proline. This appears to make little difference to the hydrophobic staple effect, since the contact is between the aliphatic ring atoms of proline and the aliphatic leucine component. Although the sequence of LcGST is very similar to that of AdGST1–3 (71% identity), it lacks the C-terminal helix α8 found in both the 1–3 and 1–4 isozymes. This is apparently due to the presence of residue W197 in LcGST, which stacks against helix α1. This residue overlaps with the base of helix α8 in AdGST1–3 and AdGST1–4, and precludes the last 14 residues of LcGST from forming a similar helix (Fig. 2e ▶).
Fig. 3.

Structure-based sequence alignment of insect GSTs. Secondary structure is indicated above the sequences in red. The kink in helix α4 is indicated (Θ). The hydrophobic staple at the base of helix α6 is indicated (<>), as is the N-terminal capping m ([]). Regions of high structural conservation are boxed with amino acids indicated in bold upper case. Residue numbers are those for AdGST1–4. The region of AdGST1–4 with no electron density (around helix α2) is indicated with a horizontal line. Helix α8, unique to the AdGST isozymes is indicated. The helix observed linking the N- and C-terminal domains of AdGST1–4 is indicated as α3B. G-site residues have shaded backgrounds. H-site residues have white letters on a black background.
The dimer interface of the GSTs forms a V-shape cleft lined primarily with residues from helix α4 and α5 in both monomers and features extensive hydrophobic and hydrogen bonding interactions. The first of these, helix α4, has a bulge in the middle that widens the cleft in the dimer interface at its entrance. Although the bulge is found in all three delta-GST structures, the dimer interface of AdGST1–4 is much less accessible because of the presence of the relatively bulky residues E116 and R134 on helix α4. (The equivalent AdGST1–3 residues are D110 and N126). Consequently, it appears unlikely that AdGST1–4 can bind ligands in this interface in a manner observed in other GSTs such as the schistosomal mu-class GST (McTigue et al. 1995; Ji et al. 1996). This observation may explain kinetic data indicating that pyrethroid is a competitive inhibitor for AdGST1–4 and noncompetitive for AdGST1–3 (Jirajaroenrat et al. 2001). This compound could bind in the dimer interface of AdGST1–3 but is unlikely to do so in AdGST1–4.
The residues forming the putative H-site are the same between the AdGST isozymes. This was unexpected because of the different kinetics for the hydrophobic cosubstrate 1-chloro-2,4-dinitrobenzene (CDNB) for the two isozymes (Table 1). H-site residues were ideied as those forming the hydrophobic cavity next to the GSH binding site. For AdGST1–3 (AdGST1–4) they are Y105 (Y111), Y113 (Y119), F117 (F123), F203 (F212), and Y206 (Y215) (Figs. 1b,c and 2d). The first three of these residues are equivalent to those found in LcGST. The additional H-site residue, F203 (F212), together with Y105 (Y111) and Y113 (Y119) form a small pocket next to the GSH sulfhydryl group. In AdGST1–3, this pocket is occupied by a single water molecule that forms H-bond interactions with Y105Oγ (3.8 Å) and Y113Oγ (2.9 Å) and is 5.0 Å from the GSH sulfhydryl group. (No equivalent water molecule is found in the LcGST structure or the AdGST1–4 structure. However, care should be taken in interpreting this result because of the different resolutions of these structures). Residue Y113 in LcGST (Y113 in AdGST1–3) was initially hypothesized to be involved in GSH activation, but site-directed mutagenesis has shown it to be relatively unimportant in this regard (Caccuri et al. 1997). When the amino acid sequences of AdGST1–3 and AdGST1–4 are aligned with the other GSTs from the alternately spliced gene family (AdGST1–1 and AdGST1–2) (data not shown), it is observed that the H-site residues on helix α3 are identical, and the residues on helix α8 vary. In the 1–3 (and 1–4) isozymes, the residues are F203 (F212) and Y206 (Y215). In AdGST1–2, the equivalent residues are A208 and F211, and in the 1–1 isozyme they are F203 and K206. Thus, structural differences in the H-site between the isozymes has been generated by mutations of helix α8.
Table 1.
Kinetic parameters of AdGST1-3 and AdGST1-4
| Kinetic parameters | AdGST1-3 | AdGST1-4 |
| Vmax | 67.5 ± 1.97 | 40.3 ± 1.89 |
| Km CDNB | 0.100 ± 0.012 | 0.523 ± 0.067 |
| Km GSH | 0.404 ± 0.054 | 0.833 ± 0.084 |
| kcat | 26.9 | 16.9 |
| kcat/Km CDNB | 269 | 32 |
| kcat/Km GSH | 67 | 20 |
Adapted from Jirajaroenrat et al. (2001). The units are: Vmax, μmole/min/mg° Km, mM, kcat, s−1; kcat/Km, mM−1s−1. The data are the mean ± standard error of at least three separate experiments.
Comparison of the AdGST structures
A crucial difference between the structures of the two AdGST isozymes is the presence and absence of GSH. AdGST1–3 has glutathione bound in a fashion similar to LcGST (Fig. 1a ▶). The absence of GSH in AdGST1–4 has had some intriguing effects. In this structure, no electron density was observed for residues 38 to 49 in either monomer. Two water molecules were observed in the G-site, where GSH is observed to bind in AdGST1–3 and LcGST. The side chains of residues N32, M34, H38, H50, and L52 in AdGST1–4 appear to be disordered. Of these residues, H38, H50, and L52 are G-site residues. The main-chain carbonyl oxygen and nitrogen atoms of L52 accept and donate hydrogen bonds (respectively) to GSH (Fig. 1a ▶). These observations strongly indicate that an induced fit mechanism operates in delta-class GSTs. Direct structural evidence of an induced fit mechanism was found previously for alpha- (Cameron et al. 1995), phi- (Neuefeind et al. 1997b), and pi-class GSTs (Oakley et al. 1998). This feature of AdGST1–4 is analogous to that observed in pi- and phi-class GSTs (Neuefeind et al. 1997b; Oakley et al. 1998). The alpha-class GST is different in that the C-terminal alpha helix (which normally buries the H-site) is disordered in the absence of hydrophobic cosubstrate. This feature of the enzyme may contribute to the removal of GSH-conjugate products from the active site, as increasing disorder is entropically favorable.
Helices α8 in AdGST1–3 and AdGST1–4 align well at the N-terminal end, but diverge as the C-terminal is approached (Fig. 2f ▶). The crystallographic data indicate that this helix is inherently flexible, since the B-factors increase with proximity to the C-terminus (In AdGST1–4, B-factors range from 35 Å2 at G201 to 60 Å2 at Y215; in AdGST1–3 they range from 11 Å2 to 50 Å2 in equivalent residues). Helix α8 in AdGST1–4 has swung 5.15° closer to the center of the dimer interface relative to AdGST1–3 (Fig. 2f ▶). This results in shifts of 2.08 Å in residue F212 and a 2.1 Å shift in residue Y215 in AdGST1–4 relative to the equivalent AdGST1–3 residues. The movements in helix α8 cause a general shift in the putative H-site residues on helix α3 (Fig. 2d ▶). In particular, Y119 (AdGST1–4) has shifted 2.0 Å toward the G-site relative to Y113 (AdGST1–4). Furthermore, Y111 (AdGST1–4) is shifted 1.45 Å toward the G-site compared to Y105 (AdGST1–3) (Fig. 2d ▶). The differences in the location of helix α8 and H-site residues between AdGST1–3 and AdGST1–4 may be isozyme-specific and may be responsible for the differences in catalytic properties for CDNB (Table 1). Another explanation of the differences in the H-site configuration of AdGST1–3 and AdGST1–4 is the presence and absence of GSH. Although GSH maintains no contacts with H-site residues, residue M34 (the side chain of which is disordered in AdGST1–4) is observed to be 5.9 Å from helix α8 in AdGST1–3. In AdGST1–4, there appears to be a mutual movement of residues in helix α8 and the flexible loop away from each other (Fig. 2f ▶). The Cα to Cα distances from L33 to Y215 (AdGST1–4) and from L33 to Y206 (AdGST1–3) are 14.84 and 13.61 Å, respectively.
Comparisons with LcGST
LcGST superimposes with AdGST1–3 and AdGST1–4 with root mean square deviations (RMSDs) of 0.81 Å (over 194 Cα atoms out of 207 amino acids) and 0.96 Å (over 190 Cα atoms out of 217 amino acids), respectively. The two AdGST structures superimpose with an RMSD of 1.05 Å over 207 Cα atoms out of 217 amino acids. Since the Lucilia GST sequence has greater similarity to AdGST1–3, the lower RMSD for this comparison is to be expected. This number is deceptive however, because several active-site residues from Lucilia GST align more closely with those from AdGST1–4 than with AdGST1–3 (see below).
AdGST1–3 and Lucilia GST sequences align with no gaps, while AdGST1–4 aligns with three inserts (Fig. 3 ▶). The first major insert, five residues, results in the formation of a small helix located between helices α3 and α4 in sequence, linking the two domains of the protein (Fig. 2b ▶). This results in a bulge at the base of the dimer interface. This insert does not appear to affect the active site directly. The other inserts are located between helices α4 and α5 (two residues), extending the intervening loop, in the loop connecting strands β3 and β4 (one residue), and in the loop connecting helices α5 and α6 (one residue). These inserts, located exclusively in loops connecting elements of secondary structure, do not appear to affect the topology of the active site. However, when kinetic characterization studies were performed, the two GSTs possessed quite different catalytic properties (Table 1). This was also observed for other substrates as well as in inhibition experiments. In a determination of inhibition kinetics using the pyrethroid insecticide permethrin, AdGST1–3 displayed noncompetitive inhibition, whereas AdGST1–4 showed competitive inhibition (Jirajaroenrat et al. 2001). The experimental data demonstrate that although the two proteins appear to possess the same active-site topology, the enzymatic characteristics exhibited are significantly different. This may be due to the aforementioned differences in the composition of the dimer interface (a potential ligand binding site) in AdGST1–3 and AdGST1–4.
Unexpectedly, the H-site residues of LcGST (with GSH bound) align more closely to AdGST1–4 than to AdGST1–3. This is despite having higher sequence identity with AdGST1–3 and also having GSH bound (versus the AdGST1–4 in apo form). The distance from Y113Oγ (LcGST) to Y119Oγ (AdGST1–4) is only 0.77 Å, compared to 2.01 Å for the Y113 (AdGST1–3) to Y113 (LcGST) distance. Similarly, the Oγ distance for Y111 (AdGST1–4) to Y105 (LcGST) is 0.54 Å compared to 1.45 Å for the equivalent Y105 (AdGST1–3) to Y105 (LcGST) distance. This result lends further evidence to the above suggestion that the H-site residue configuration found in AdGST1–4 is isozyme-specific and not determined by whether GSH is bound to the enzyme.
Alternate splicing
AdGST1–3 and AdGST1–4 result from alternate splicing in a single gene (Pongjaroenkit et al. 2001). Consequently, residues 1–45 in both enzymes result from the same exon. These residues correspond to the N-terminal GSH binding domain up to the middle of helix α2, and include several GSH binding residues. This modular gene structure allows isozyme heterogeneity by allowing for different C-terminal domains. It is the C-terminal domain that contains the H-site residues, and thus different substrate specificity could be generated efficiently by this gene arrangement. However, the gene arrangement must place some constraints on sequence diversity in the C-terminal domains: they must be able to form the correct fold and structurally compliment the N-terminal domain. Investigation of the contacts between residues 1–45 and the rest of the enzymes reveals that the contacting residues are almost identical. The exceptions are residue C69 (AdGST1–3) versus Q70 (AdGST1–4) in the middle of helix α3, and residues I195 (AdGST1–3) versus K204 (AdGST1–4) at the base of helix α8. These residues show very similar packing and do not appear to affect the overall fold.
Materials and methods
The cloning, purification, crystallization, and data collection methods have been reported (Jirajaroenrat et al. 2001; Oakley et al. 2001). Briefly, crystals of GST1–3 were obtained by the hanging drop technique using 0.2 M ammonium acetate, 0.1 M sodium acetate pH 4.6, 30% PEG 4,000 as the mother liquor at room temperature. Crystals of AdGST1–4 were obtained by the hanging drop technique with 0.2 M sodium acetate, 30% PEG 8,000, 0.1 M sodium cacodylate pH 6.5 as the mother liquor at room temperature. Data were collected using a Mar Research area detector mounted with Ni-focusing mirrors on an RU-200 X-ray generator producing Cu Kα radiation.
Structure solution and refinement
The AdGST1–4 crystals were in the space group P3221, with unit cell a=b=49.5, c=271.86, α=β=90°, γ=120°. This gives a Matthews' coefficient for AdGST1–4 of 1.93 Å3/dalton for an asymmetric unit containing 1 dimer. The structure was determined using molecular replacement in the MOLREP program (Vagin and Teplyakov 2000) and coordinates from the dimeric LcGST structure (Wilce et al. 1995) as the search model. The optimal solution corresponded to the top peak in both rotation and translation functions. Statistics for the solutions are given in Table 2. The solution was confirmed by the observation of the C-terminal helix, not found in Lucilia GST, in the initial electron density maps calculated from this model. This initial model had an R factor and Rfree of 45.9% and 45.2%, respectively (5% of reflections, chosen randomly, were selected for the Rfree set). The R factor (and Rfree) reduced to 43.3% (41.0%) after rigid body refinement. The model was then subject to three cycles of rebuilding using σa-weighted 2fo-fc and fo-fc maps in O (Jones et al. 1991), and positional and individual restrained B-factor refinement in CNS (Brünger et al. 1998). NCS restraints were used throughout the refinement. Map interpretation was aided by twofold NCS averaging using the RAVE package (Kleywegt and Read 1997). No σ or resolution cutoffs were used in refinement; consequently, bulk solvent correction was used in refinements. After initial refinement, the C-terminal helix was built into the model (R factor = 28%, Rfree=30.0%). Five more cycles of rebuilding and positional and individual restrained B-factor refinement were performed, and 40 water molecules were built into the electron density maps. Electron density for NCS-related residues were compared and where they deviated, NCS restraints were released. No electron density for GSH was observed in the maps. No electron density was observed for residues 38 to 49 in either monomer. However, when these residues were removed, an increase in R factor and Rfree resulted. The final model (R factor = 22.3%, Rfree = 27.1%) includes these residues, which have very high B-factors ranging from 78 to 113 Å2. Thus, it appears that the absence of density is due to high mobility. Figure 1b ▶ shows 2fo-fc maps generated from the final model for the H-site of AdGST1–4.
Table 2.
Molecular replacement data
| AdGST1-4: | |||||
| Molecular replacement results from MOLREP using Lucilia GST as the search model | |||||
| α,β,γ | peak height (σ) | ||||
| Rotation function solution | 48.67,85.30,−53.13 | 6.96 | |||
| tX,tY,tZ | Correlation, R factor | ||||
| Translation function solution | 0.901,0576,0.488 | 47.7, 47.0 | |||
| AdGST1-3: | Molecular replacement results from AMORE using AdGST1-4 as the search model | ||||
| α,β,γ | peak height (σ) | ||||
| Rotation function solution | 45.21,89.82,158.29 | 10.0 | |||
| tX,tY,tZ | Correlation, R factor | ||||
| Translation function solution 1 | 0.805,0.305,0.000 | 20.7 64.0 | |||
| Translation function solution 2 | 0.807,0.305,0.666 | 52.9 53.9 | |||
| Translation function solution 3 | 0.806,0.305,0.333 | 65.2 66.6 | |||
The AdGST1–3 crystals were tetragonal (a=b=87.81, Å c=166.1 Å, α=β=γ=90°). The space group was P43. With this symmetry, the asymmetric unit contains three dimers with a Matthews' coefficient of 2.24 Å3/dalton or four dimers with a Matthews' coefficient of 1.68 Å3/dalton. Attempts to solve the structure using MOLREP and the final AdGST1–4 model were unsuccessful. AMORE (CCP4 1994) was then used, because of its ability to subtly control the search criteria. Strong peaks were found in the rotation function for the 4/m symmetry space groups. Using the AdGST1–4 dimer as a search model, a 10.0 σ peak was found in the rotation function. The highest nonsolution peak was 4.3 σ. The first peak found in the translation function had a correlation coefficient of 20.7% and an R factor of 64.0%. This solution was fixed, and another peak, using the same rotation solution, was found (correlation coefficient = 52.9%, R factor = 53.9%). This solution was in turn fixed and another peak was found (correlation coefficient = 65.2%, R factor = 66.6%). The solutions in the translation function corresponded to similar positions with translations equivalent to 0, 1/3, and 2/3 of the unit cell along the Z direction. Full statistics for the solutions are given in Table 2. The packing gives three dimers, aligned along the Z axis. The crystal packing is pseudo-P43212. The packing is also pseudo P41 with the transformation c' = 1/3c. The solution was confirmed by the observation of electron density for GSH (not in the model) in the active site. The initial model had an R factor of 69.7% (Rfree = 66.9%). This rapidly reduced to 58.3% (Rfree = 59.5%) after rigid body refinement. After three cycles of building (using σa-weighted 2fo-fc and fo-fc maps), NCS restrained positional and restrained individual atom B-factor refinement, the R factor reduced to 37.3% (Rfree = 41.9%). Maps were improved with sixfold NCS averaging. At this stage, NCS restraints were released, resulting in a slight decrease in R factor (35.7%) and Rfree (35.7%) after another cycle of positional and B-factor refinement. After another two rounds of energy minimization and B-factor refinement, the R factor reduced to 30.0% (Rfree = 33.8%). At this stage, numerous peaks in fo-fc and 2fo-fc maps corresponding to water molecules were observed. Twenty cycles of ARP/Refmac refinement (Lamzin and Wilson 1993) were used to automatically build and refine water molecules in the model. The resulting model had 641 waters and an R factor of 23.3% (Rfree = 29.9%). This model was subjected to another five rounds of rebuilding and positional and B-factor refinement, during which six GSH molecules were built into the model. The 2fo-fc maps generated from the final model are shown for the H-site of AdGST1–3 (Fig. 1c ▶). The electron density for GSH is shown in Figure 1a ▶.
Structure analysis
Structure alignment and RMSD calculations were performed using LSQMAN (Kleywegt 1999). The individual and aligned structures are shown in Figure 2a ▶–c. The geometry of the structures was analyzed with PROCHECK (Laskowski et al. 1993) and CNS. The final models have excellent statistics (Table 3). The two AdGST structures were aligned with the LcGST structure in the STAMP structure-based sequence alignment program (Russell and Barton 1992). The resulting alignment is shown in Figure 3 ▶.
Table 3.
Refinement statisticsa
| AdGST1-4 | AdGST1-3 | |
| Number of atoms | ||
| (protein) | 3494 | 9960 |
| (water) | 40 | 641 |
| (GSH) | — | 120 |
| Rfactor | 22.3 (31.1) | 22.3 (28.1) |
| Rfree | 27.1 (37.8) | 23.1 (28.4) |
| Resolution range (Å) | 19.92–2.45 | 17.22–1.75 |
| Reflections in working set | 14264 (1996) | 91965 (17355) |
| Completeness (%) | 94.4 (85.7) | 77.2 (87.1) |
| Reflections in test set | 725 (125) | 4830 (878) |
| Completeness (%) | 5.1 | 5.0 |
| RMS deviation from ideal geometry | ||
| Bond lengths (Å) | 0.007 | 0.006 |
| Bond angles (°) | 1.3 | 1.2 |
| Dihedral angles (°) | 21.4 | 20.7 |
| Improper angles (°) | 0.81 | 0.81 |
| RMSD on Bonded Bs (Å2) | 2.7 | 2.0 |
| Residues in most favorable region | ||
| Of ramachandran plot (%) | 89.1 | 91.3 |
a Numbers in parentheses refer to the highest resolution bin (1.75–1.86 for AdGST1-3, 2.45–2.60 for AdGST1-4).
Accession numbers
AdGST1–3 and AdGST1–4 coordinates have been deposited with the Protein Data Bank with ID codes 1JLV and 1JLW, respectively.
Acknowledgments
This research is supported by a grant from The Thailand Research Fund (A.J.K.) and a UWA/ARC Small Research Grant (A.J.O.). A.J.O. is a recipient of a UWA postdoctoral research fellowship and ARC fellowship. M.C.J.W. is supported by a Raine Medical Research Foundation Fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
GST, glutathione S-transferase
GSH, glutathione
Ad, Anopheles dirus species B
Lc, Lucilia cuprina
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.21201.
References
- Board, P., Baker, R.T., Chelvanayagam, G., and Jermiin, L.S. 1997. Zeta, a novel class of glutathione transferases in a range of species from plants to humans. Biochem. J. 328 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Board, P.G., Coggan, M., Chelvanayagam, G., Easteal, S., Jermiin, L.S., Schulte, G.K., Danley, D.E., Hoth, L.R., Griffor, M.C., Kamath, et al. 2000. Ideication, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 275 24798–24806. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, N., Pannu, N.S., Read, et al. 1998. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Caccuri, A.M., Antonini, G., Nicotra, M., Battistoni, A., Bello, M.L., Board, P.G., Parker, M.W., and Ricci, G. 1997. Catalytic mechanism and role of hydroxyl residues in the active site of theta class glutathione S-transferases. Investigation of Ser-9 and Tyr-113 in a glutathione S-transferase from the Australian sheep blowfly, Lucilia cuprina. J. Biol. Chem. 272 29681–29686. [DOI] [PubMed] [Google Scholar]
- Cameron, A.D., Sinning, I., L'Hermite, G., Olin, B., Board, P.G., Mannervik, B., and Jones, T.A. 1995. Structural analysis of human alpha-class glutathione transferase A1–1 in the apo-form and in complexes with ethacrynic acid and its glutathione conjugate. Structure 3 717–727. [DOI] [PubMed] [Google Scholar]
- Clark, A.G. and Dauterman, W.C., 1982. The characterization by affinity chromatography of glutathione S-transferases from different strains of house fly. Pestic. Biochem. Physiol. 17 307–314. [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Dirr, H., Reinemer, P., and Huber, R. 1994. Refined crystal structure of porcine class Pi glutathione S-transferase (pGST P1–1) at 2.1 A resolution. J. Mol. Biol. 243:72–92. [DOI] [PubMed] [Google Scholar]
- Dragani, B., Stenberg, G., Melino, S., Petruzzelli, R., Mannervik, B., and Aceto, A. 1997. The conserved N-capping box in the hydrophobic core of glutathione S-transferase P1–1 is essential for refolding: Ideication of a buried and conserved hydrogen bond important for protein stability. J. Biol. Chem. 272 25518–25523. [DOI] [PubMed] [Google Scholar]
- Dulhunty, A., Gage, P., Curtis, S., Chelvanayagam, G., and Board, P. 2001. The glutathione transferase structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J. Biol. Chem. 276 3319–3323. [DOI] [PubMed] [Google Scholar]
- Garcia-Saez, I., Parraga, A., Phillips, M.F., Mantle, T.J., and Coll, M. 1994. Molecular structure at 1.8 Å of mouse liver class pi glutathione S-transferase complexed with S-(p-nitrobenzyl)glutathione and other inhibitors. J. Mol. Biol. 237 298–314. [DOI] [PubMed] [Google Scholar]
- Hayes, J.D. and Pulford, D.J. 1995. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 30 445–600. [DOI] [PubMed] [Google Scholar]
- Ji, X., Zhang, P., Armstrong, R.N., and Gilliland, G.L. 1992. The three-dimensional structure of a glutathione S-transferase from the mu gene class. Structural analysis of the binary complex of isoenzyme 3–3 and glutathione at 2.2-Å resolution. Biochemistry 31 10169–10184. [DOI] [PubMed] [Google Scholar]
- Ji, X., Armstrong, R.N., and Gilliland, G.L. 1993. Snapshots along the reaction coordinate of an SNAr reaction catalyzed by glutathione transferase. Biochemistry 32 12949–12954. [DOI] [PubMed] [Google Scholar]
- Ji, X., Johnson, W.W., Sesay, M.A., Dickert, L., Prasad, S.M., Ammon, H.L., Armstrong, R.N., and Gilliland, G.L. 1994. Structure and function of the xenobiotic substrate binding site of a glutathione S-transferase as revealed by X-ray crystallographic analysis of product complexes with the diastereomers of 9–(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthrene. Biochemistry 33 1043–1052. [DOI] [PubMed] [Google Scholar]
- Ji, X., von Rosenvinge, E.C., Johnson, W.W., Tomarev, S.I., Piatigorsky, J., Armstrong, R.N., and Gilliland, G.L. 1995. Three-dimensional structure, catalytic properties, and evolution of a sigma class glutathione transferase from squid, a progenitor of the lens S-crystallins of cephalopods. Biochemistry 34 5317–5328. [DOI] [PubMed] [Google Scholar]
- Ji, X., von Rosenvinge, E.C., Johnson, W.W., Armstrong, R.N., and Gilliland, G.L. 1996. Location of a potential transport binding site in a sigma class glutathione transferase by X-ray crystallography. Proc. Natl. Acad. Sci. 93 8208–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, X., Blaszczyk, J., Xiao, B., O'Donnell, R., Hu, X., Herzog, C., Singh, S.V., and Zimniak, P. 1999. Structure and function of residue 104 and water molecules in the xenobiotic substrate-binding site in human glutathione S-transferase P1–1. Biochemistry 38 10231–10238. [DOI] [PubMed] [Google Scholar]
- Jirajaroenrat, K., Pongjaroenkit, S., Krittanai, C., Prapanthadara, L., and Ketterman, AJ. 2001. Heterologous expression and characterization of alternatively spliced glutathione S-transferases from a single Anopheles gene. Insect Biochem. Mol. Biol. 31 867–875. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kanaoka, Y., Ago, H., Inagaki, E., Nanayama, T., Miyano, M., Kikuno, R., Fujii, Y., Eguchi, N., Toh, H., Urade, Y., et al. 1997. Cloning and crystal structure of hematopoietic prostaglandin D synthase. Cell 90 1085–1095. [DOI] [PubMed] [Google Scholar]
- Kanaoka, Y., Fujimori, K., Kikuno, R., Sakaguchi, Y., Urade, Y., and Hayaishi, O. 2000. Structure and chromosomal localization of human and mouse genes for hematopoietic prostaglandin D synthase. Conservation of the ancestral genomic structure of sigma-class glutathione S-transferase. Eur. J. Biochem. 267 3315–3322. [DOI] [PubMed] [Google Scholar]
- Ketterman, A.J., Prommeenate, P., Boonchauy, C., Chanama, U., Leetachewa, S., Promtet, N., and Prapanthadara, L. 2001. Single amino acid changes outside the active site significantly affect activity of glutathione S-transferases. Insect. Biochem. Mol. Biol. 31 65–74. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. 1999. Experimental assessment of differences between related protein crystal structures. Acta Crystallogr. D Biol. Crystallogr. 55 1878–1884. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J. and Read, R.J. 1997. Not your average density. Structure 5 1557–1569. [DOI] [PubMed] [Google Scholar]
- Lamzin, V.S. and Wilson, K.S. 1993. Automated refinement of protein models. Acta Crystallogr. D Biol. Crystallogr. 49 129–147. [DOI] [PubMed] [Google Scholar]
- Lamoureux G.L. and Rusness, D.G. 1989. The role of glutathione and glutathione-S-transferases in pesticide metabolism, selectivity, and mode of action in plants and insects. In Glutathione. chemical, biochemical, and medical aspects. Part B. (eds. D. Dolphin, O. Avroamovic, and R. Poulson), pp. 153–196. John Wiley & Sons, New York, NY.
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. App. Crystallog. 26 283–291. [Google Scholar]
- Mannervik, B., Awasthi, Y.C., Board, P.G., Hayes, J.D., Di Ilio, C., Ketterer, B., Listowsky, I., Morgenstern, R., Muramatsu, M., Pearson, et al. 1992. Nomenclature for human glutathione transferases. Biochem. J. 282 305–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue, M.A., Williams, D.R., and Tainer, J.A. 1995. Crystal structures of a schistosomal drug and vaccine target: Glutathione S-transferase from Schistosoma japonica and its complex with the leading antischistosomal drug praziquantel. J. Mol. Biol. 246 21–27. [DOI] [PubMed] [Google Scholar]
- Neuefeind, T., Huber, R., Dasenbrock, H., Prade, L., and Bieseler, B. 1997b. Crystal structure of herbicide-detoxifying maize glutathione S-transferase-I in complex with lactoylglutathione: Evidence for an induced-fit mechanism. J. Mol. Biol. 274 446–453. [DOI] [PubMed] [Google Scholar]
- Neuefeind, T., Huber, R., Reinemer, P., Knablein, J., Prade, L., Mann, K., Bieseler, and B. 1997a. Cloning, sequencing, crystallization and X-ray structure of glutathione S-transferase-III from Zea mays var. mutin: A leading enzyme in detoxification of maize herbicides. J. Mol. Biol. 274 577–587. [DOI] [PubMed] [Google Scholar]
- Nordstrand, K., Åslund, F., Holmgren, A., Otting, G., and Berndt, K.D. 1999. NMR structure of Escherichia coli glutaredoxin 3-glutathione mixed disulfide complex: Implications for the enzymatic mechanism. J. Mol. Biol. 286 541–552. [DOI] [PubMed] [Google Scholar]
- Oakley, A.J., Lo Bello, M.L., Battistoni, A., Ricci, G., Rossjohn, J., Villar, H.O., and Parker, M.W. 1997. The structures of human glutathione transferase P1–1 in complex with glutathione and various inhibitors at high resolution J. Mol. Biol. 274 84–100. [DOI] [PubMed] [Google Scholar]
- Oakley, A.J., Lo Bello, M., Ricci, G., Federici, G., and Parker, M.W. 1998. Evidence for an induced-fit mechanism operating in pi class glutathione transferases. Biochemistry 37 9912–9917. [DOI] [PubMed] [Google Scholar]
- Oakley, A.J., Lo Bello, M., Nuccetelli, M., Mazzetti, A.P., and Parker, M.W. 1999. The ligandin (non-substrate) binding site of human Pi class glutathione transferase is located in the electrophile binding site (H-site). J. Mol. Biol. 291 913–926. [DOI] [PubMed] [Google Scholar]
- Oakley, A.J., Jirajaroenrat, K., Harnnoi, T., Ketterman, A.J., Wilce, M.C. 2001. Crystallization of two glutathione S-transferases from an unusual gene family. Acta Crystallogr. D Biol. Crystallogr. 57 870–872. [DOI] [PubMed] [Google Scholar]
- Pemble, S.E. and Taylor, J.B. 1992. An evolutionary perspective on glutathione transferases inferred from class-Theta glutathione transferase cDNA sequences. Biochem. J. 287 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemble, S.E., Wardle, A.F., and Taylor, J.B. 1996. Glutathione S-transferase class kappa: Characterization by the cloning of rat mitochondria GST and ideication of a human homologue. Biochem. J. 319 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, R.S. 2001.Current status of malaria and potential for control. Clin. Microbiol. Rev. 14 208–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongjaroenkit, S., Jirajaroenrat, K., Boonchauy, C., Chanama, U., Leetachewa, S., Prapanthadara, L., and Ketterman, A.J. 2001. Genomic organization and putative promoters of highly conserved glutathione S-transferases originating by alternative splicing in Anopheles dirus. Insect. Biochem. Mol. Biol. 31 75–85. [DOI] [PubMed] [Google Scholar]
- Prade, L., Huber, R., Manoharan, T.H., Fahl, W.E., and Reuter, W. 1997. Structures of class pi glutathione S-transferase from human placenta in complex with substrate, transition-state analogue and inhibitor. Structure 5 1287–1295. [DOI] [PubMed] [Google Scholar]
- Prapanthadara, L.A., Hemingway, J., and Ketterman, A.J. 1993. Partial purification and characterization of glutathione S-transferases involved in DDT resistance from the mosquito Anopheles gambiae. Pestic. Biochem. Physiol. 47 119–133. [Google Scholar]
- Prapanthadara, L., Ketterman, A.J., and Hemingway, J. 1995. DDT-resistance in Anopheles gambiae Giles from Zanzibar Tanzania based on increased DDT-dehydrochlorinase activity of glutathione S-transferases. Bull. Entomol. Res. 85 267–274. [Google Scholar]
- Prapanthadara, L., Promtet, N., Koottathep, S., Somboon, P., and Ketterman, A.J. 2000. Isoenzymes of glutathione S-transferase from the mosquito Anopheles dirus species B: The purification, partial characterization and interaction with various insecticides. Insect Biochem. Mol. Biol. 30 395–403. [DOI] [PubMed] [Google Scholar]
- Raghunathan, S., Chandross, R.J., Kretsinger, R.H., Allison, T.J., Penington, C.J., and Rule GS. 1994. Crystal structure of human class mu glutathione transferase GSTM2–2. Effects of lattice packing on conformational heterogeneity. J. Mol. Biol. 238 815–832. [DOI] [PubMed] [Google Scholar]
- Reinemer, P., Dirr, H.W., Ladenstein, R., Huber, R., Lo Bello, M., Federici, G., and Parker, M.W. 1992. Three-dimensional structure of class pi glutathione S-transferase from human placenta in complex with S-hexylglutathione at 2.8 A resolution. J. Mol. Biol. 227 214–226. [DOI] [PubMed] [Google Scholar]
- Reinemer, P., Prade, L., Hof, P., Neuefeind, T., Huber, R., Zettl, R., Palme, K., Schell, J., Koelln, I., Bartunik, H.D., et al. 1996. Three-dimensional structure of glutathione S-transferase from Arabidopsis thaliana at 2.2 Å resolution: Structural characterization of herbicide-conjugating plant glutathione S-transferases and a novel active site architecture. J. Mol. Biol. 255 289–309. [DOI] [PubMed] [Google Scholar]
- Rossjohn, J., McKinstry, W.J., Oakley, A.J., Verger, D., Flanagan, J., Chelvanayagam, G., Tan, K.L., Board, P.G., and Parker, M.W. 1998a. Human theta class glutathione transferase: The crystal structure reveals a sulfate-binding pocket within a buried active site. Structure 6 309–322. [DOI] [PubMed] [Google Scholar]
- Rossjohn, J., Polekhina, G., Feil, S.C., Allocati, N., Masulli, M., De Illio, C., and Parker, M.W. 1998b. A mixed disulfide bond in bacterial glutathione transferase: Functional and evolutionary implications. Structure 6 721–734. [DOI] [PubMed] [Google Scholar]
- Russell, R.B. and Barton, G.S. 1992. Multiple protein sequence alignment from tertiary structure comparison. Proteins 14 309–323. [DOI] [PubMed] [Google Scholar]
- Sinning, I., Kleywegt, G.J., Cowan, S.W., Reinemer, P., Dirr, H.W., Huber, R., Gilliland, G.L., Armstrong, R.N., Ji, X., Board, P.G., et al. 1993. Structure determination and refinement of human alpha class glutathione transferase A1–1, and a comparison with the Mu and Pi class enzymes. J. Mol. Biol. 232 192–212. [DOI] [PubMed] [Google Scholar]
- Sundquist, A.R. and Fahey, R.C. 1989. The function of gamma-glutamylcysteine and bis-gamma-glutamylcystine reductase in Halobacterium halobium. J. Biol. Chem. 264 719–725. [PubMed] [Google Scholar]
- Snyder, M.J., and Maddison, D.R. 1997. Molecular phylogeny of glutathione-S-transferases. DNA Cell Biol. 16 1373–1384. [DOI] [PubMed] [Google Scholar]
- Stenberg, G., Dragani, B., Cocco, R., Mannervik, B., and Aceto, A. 2000. A conserved "hydrophobic staple m" plays a crucial role in the refolding of human glutathione transferase P1–1. J. Biol. Chem. 275 10421–10428. [DOI] [PubMed] [Google Scholar]
- Trigg, P.I. and Kondrachine, A.V. 1998. Commentary: Malaria control in the 1990s. Bull. World Health Organ. 76 11–16. [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. and Teplyakov, A. 2000. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 56 1622–1624. [DOI] [PubMed] [Google Scholar]
- Vega, M.C., Walsh, S.B., Mantle, T.J., and Coll, M. 1998. The three-dimensional structure of Cys-47-modified mouse liver glutathione S-transferase P1–1. Carboxymethylation dramatically decreases the affinity for glutathione and is associated with a loss of electron density in the alphaB-310B region. J. Biol. Chem. 273 2844–2850. [DOI] [PubMed] [Google Scholar]
- Wilce, M.C., Board, P.G., Feil, S.C., Parker, M.W. 1995. Crystal structure of a theta-class glutathione transferase. EMBO J. 14 2133–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilce, M.C. and Parker, M.W. 1994. Structure and function of glutathione S-transferases. Biochim Biophys Acta. 1205 1–18. [DOI] [PubMed] [Google Scholar]
- Xiao, B., Singh, S.P., Nanduri, B., Awasthi, Y.C., Zimniak, P., and Ji, X. 1999. Crystal structure of a murine glutathione S-transferase in complex with a glutathione conjugate of 4-hydroxynon-2-enal in one subunit and glutathione in the other: Evidence of signalling across the dimer interface. Biochemistry 38 11887–11894. [DOI] [PubMed] [Google Scholar]