

Abstract
The monomeric peptide methionine sulfoxide reductase (MsrA) catalyzes the irreversible thioredoxin-dependent reduction of methionine sulfoxide. The crystal structure of MsrAs from Escherichia coli and Bos taurus can be described as a central core of about 140 amino acids that contains the active site. The core is wrapped by two long N- and C-terminal extended chains. The catalytic mechanism of the E. coli enzyme has been recently postulated to take place through formation of a sulfenic acid intermediate, followed by reduction of the intermediate via intrathiol-disulfide exchanges and thioredoxin oxidation. In the present work, truncated MsrAs at the N- or C-terminal end or at both were produced as folded entities. All forms are able to reduce methionine sulfoxide in the presence of dithiothreitol. However, only the N-terminal truncated form, which possesses the two cysteines located at the C-terminus, reduces the sulfenic acid intermediate in a thioredoxin-dependent manner. The wild type displays a ping-pong mechanism with either thioredoxin or dithiothreitol as reductant. Kinetic saturation is only observed with thioredoxin with a low KM value of 10 μM. Thus, thioredoxin is likely the reductant in vivo. Truncations do not significantly modify the kinetic properties, except for the double truncated form, which displays a 17-fold decrease in kcat/KMetSO. Alternative mechanisms for sulfenic acid reduction are also presented based on analysis of available MsrA sequences.
Keywords: Methionine sulfoxide reductase, truncated enzymes, sulfenic acid reduction, thioredoxin
The peptide methionine sulfoxide reductase (MsrA) is a ubiquitous enzyme that catalyzes the irreversible reduction of both free and protein-bound methionine sulfoxide (MetSO) (Moskovitz et al. 1995, 1996). In vivo, its role is described as essential in protecting cells against oxidative stress by restoring the biological properties of the targeted proteins (Abrams et al. 1981; Moskovitz et al. 1997, 1998; Sun et al. 1999). In addition to their protective function, MsrAs have been postulated to play a role in infection of cells by pathogenic bacteria (Wizemann et al. 1996; El Hassouni et al. 1999). Recently, two MsrAs from Escherichia coli and Bos taurus have been extensively studied at both the mechanistic and structural levels (Boschi-Muller et al. 2000; Lowther et al. 2000a,b; Tête-Favier et al. 2000). In both cases, three cysteine residues (numbering 51,198 and 206 in E. coli MsrA) were shown to be involved in the catalytic mechanism. In the E. coli enzyme, the data supported a three-step chemical mechanism in vivo in agreement with (1) a nucleophilic attack of the essential Cys-51 towards the sulfur atom of the sulfoxide substrate leading to formation of a tetracoordinate intermediate, which rearranges into a sulfenic intermediate on Cys-51 with release of 1 mole of methionine per mole of enzyme; (2) a reduction of the sulfenic acid intermediate through thiol intradisulfide exchanges leading finally to formation of a disulfide bond between Cys-198 and Cys-206; and (3) the regeneration of the free Cys-198 and Cys-206 through a thioredoxin (Trx)-dependent recycling system process (Boschi-Muller et al. 2000). In contrast, the covalent tetracoordinate intermediate, which was also assumed to be formed in the bovine enzyme between the Cys-72 equivalent to Cys-51 and the sulfoxide of the substrate, was postulated to break down directly through thiol–disulfide exchanges with release of methionine (Lowther et al. 2000a). Therefore, although both mechanisms involve similar thiol exchanges through Cys-198 and Cys-206, they differ significantly from each other as concerns (1) the mechanism of the sulfoxide reduction, and (2) the nature of the step that leads to methionine release. The fact that the sulfenic acid intermediate was characterized for the E. coli enzyme is consistent with the generality of the sulfenic acid mechanism for all MsrAs.
The observed crystal structure of MsrAs from E. coli and Bos taurus is of the mixed α-β type, with a core containing a two-layer sandwich, α-β plaits m (Lowther et al. 2000b; Tête-Favier et al. 2000). The content of α-helices and β-strands represents less than 40% of the secondary structure. In E. coli MsrA, the central core is composed of amino acids from 43 to 182, which are wrapped by two extended chains composed of the 42 amino acids of the N-terminal end and of the 29 amino acids of the C-terminal end. The core contains the active site, which can be represented as an opened basin readily accessible to the substrate. The amino acids involved in the chemical mechanism and in the structural specificity are located within the basin. In particular, the essential Cys-51, which is oxidized to sulfenic acid during the first step of the mechanism, belongs to the almost conserved GCFWG m. The C-terminal extended chain possesses the Cys-198 and Cys-206, which are essential in the Trx-dependent recycling process. This C-terminal end also contains a glycine-rich region.
Taking into account all the structural and mechanistic data available so far, we hypothesized that truncated MsrAs at the N-terminal or the C-terminal end or at both can be isolated as folded entities, and retain the capability of reducing methionine sulfoxide, but that regeneration of Cys-51 through intervention of Trx is only achieved by the N-terminal truncated form. To validate these hypotheses, truncated MsrAs from E. coli at the N-terminal end (amino acids 1–41) or at the C-terminal end (amino acids 195–211) or at both were produced and then isolated in a pure form. The kinetic mechanism of the wild-type enzyme was determined, and the enzymatic properties of the truncated forms were studied and compared with those of the wild type. Alternative mechanisms for sulfenic acid reduction are also presented based on analysis of MsrAs primary structures.
Results
Juication of the truncations
The choice of truncations after amino acids 41 and 194 of the N-terminal and C-terminal ends of E. coli MsrA, respectively, was based on the following points.
Truncation 1–41
First, the N-terminal end has no α-helical and β-sheet structures. It makes rather limited contacts to the rest of the protein but is clearly well positioned. Second, none of the amino acids involved either in the substrate binding or in the reduction of the sulfoxide and of the sulfenic acid intermediate are located within its sequence (Boschi-Muller et al. 2000; Tête-Favier et al. 2000). Third, there exists MsrAs from Streptococcus pneumoniae and Bacillus subtilis (see Fig. 1 ▶), which possess a proteic sequence starting from or near the equivalent of E. coli Met-42, and which were described to be active (Wizemann et al. 1996; Hayes et al. 1998).
Fig. 1.

MsrAs sequence alignment of the nine active and of the putative R. capsulatus MsrAs. The sequences of nine characterized MsrAs from E. coli (Boschi-Muller et al. 2000), B. subtilis (Hayes et al. 1998), Bos taurus (Lowhter et al. 2000a), Brassica napus (abbreviated B. napus; Sadanandom et al. 1996), Erwinia chrysanthemi (E. chrys.; El Hassouni et al. 1999), Homo sapiens (H. sapiens; Kuschel et al. 1999), N. gonnhorroeae (N. gonnho; Wizemann et al. 1996), S. cerevisiae (S. cerev.; Moskovitz et al. 1997), and S. pneumoniae (S. pneumo; Wizemann et al. 1996), and of the putative R.capsulatus (R. capsu.) MsrA were aligned with Bioedit software. The residues white on black boxes are strictly conserved in the selected sequences. Black triangles indicated the position of the three catalytic Cys residues of E. coli MsrA. The Cys residues that are not conserved are indicated in white on gray boxes. α-Helices and β-strands of E. coli MsrA are indicated by gray lines and black arrows, respectively.
Truncation 195–211
The C-terminal end, composed of 29 amino acids, contains a consensus signature ExxHQxYLxK corresponding to amino acids 183–192, which are located partly within the active site, and Cys-198 and Cys-206, which are essential for the Trx-dependent recycling process. Inspection of the structure of the C-terminal end of the three molecules of MsrA observed in the asymmetric unit shows that they are quite different. Although all three molecules present no α-helical and β-sheet structures and a similar and a clearly defined positioning of residues from 183 to 192, the remaining C-terminal amino acids are either partly ordered or not, depending on the molecules considered. This C-terminal end contains a series of Gly residues between Cys-198 and Cys-206 that can render the C-terminal extended chain flexible and thus likely brings the two Cys residues near the active site Cys-51, allowing the reduction of the sulfenic intermediate (Boschi-Muller et al. 2000; Tête-Favier et al. 2000). Taking into account this structural information and the fact that reductase activity remained in mutants where Cys-198 and Cys-206 were mutated into Ser, the truncation was done after the amino acid 194.
Biochemical characterization
Wild-type and truncated MsrAs were overexpressed in E. coli strain using a vector containing the corresponding DNA MsrA sequences under the strong T7 promoter. After sonication and centrifugation, wild-type and C-truncated MsrAs were in the supernatant, whereas both N-truncated forms were in the pellet. These results support a role of the N-terminal end in the efficiency of the E. coli protein folding in vivo. However, refolding in vitro of the unsoluble forms was easily carried out after treatment with urea and elution on an anionic exchange resin. All forms of MsrAs were obtained pure as judged by SDS-PAGE gels and mass spectrometry analysis, i.e., measured mass 21533.5 ± 0.5, 18908.5 ± 0.5, and 17257.5 ± 0.5 Daltons, theoretical mass 21533, 18908, and 17257 Daltons for N-truncated, C-truncated, and double-truncated forms, respectively. DTNB reagent revealed four cysteines (Cys-51, 86, 198, and 206) and two cysteines (Cys-51 and 86) for the N-truncated form, and C-truncated and double-truncated forms, respectively, under either native or denaturating conditions (see Table 1). This is in agreement with previous results described for the wild type, which showed that all cysteines are surface residues (Boschi-Muller et al. 2000). Stoichiometry of methionine formation was determined in the absence of reductant. Two moles of methionine were formed for the N-truncated form with loss of three thiols, which is in agreement with formation of sulfenic acid on Cys-51 and of a disulfide bond between Cys-198 and Cys-206, whereas only 1 mole was found for C-truncated and double-truncated forms with loss of one thiol, which is in agreement with formation of sulfenic acid on Cys-51.
Table 1.
Stoichiometry of Met formed in the absence of reductant and free sulfhydryl content of wild-type and truncated mutant MsrAs
| No. of Cys (mole Cys/mole enzyme) | ||||||
| Decrease in free thiols | ||||||
| Enzyme | mol Met/mol enzyme | No. of Cys | ||||
| Without MetSO | With MetSOc | Calculated from the data | Theoreticalb | |||
| Wild-typea | 2.1 | 4 | 3.8 | 1 | 2.8 | 3 |
| N-truncated | 1.9 | 4 | 3.9 | 0.6 | 3.3 | 3 |
| C-truncated | 1 | 2 | 1.8 | 0.7 | 1.1 | 1 |
| N + C-truncated | 0.9 | 2 | 1.7 | 0.7 | 1.0 | 1 |
The values indicated represent the average of three independent measurements of at least two enzyme concentrations (relative SD range, 10%). Quantity of Met formed was determined by high-pressure liquid chromatography after reaction with 10 mM MetSO without any regenerating system as described under Materials and Methods section. Cysteine content was determined spectrophotometrically using DTNB under native conditions in the absence or in the presence of 10 mM MetSO as described (Materials and Methods).
a From Boschi-Muller et al. (2000).
b Based on the chemical mechanism described in Boschi-Muller et al. (2000).
c The TNB− molecule, which is released, does not significantly react with the sulfenic acid intermediate because its concentration is too low. This explains why the thiol content after MetSO treatment is 1 instead of an expected net thiol content of zero.
Determination of the kinetics parameters of wild-type and truncated MsrAs
To detect possible modifications in kcat and KM values due to truncations, detailed kinetic studies were undertaken with MetSO as a substrate and Trx or DTT as the thiol regenerating system. As shown in Figure 2 ▶, for the wild type, double reciprocal plots of initial velocity versus MetSO concentration yielded parallel slopes for different concentrations of Trx, as is typical for the "ping pong" mechanism (Fig. 2A ▶). Replotting the reciprocal Trx concentrations against the reciprocal Vmax or the reciprocal KM yielded a straight line (Fig. 2B ▶). These data can be described by the two substrate ping-pong mechanism of Equation 1, where kmax represents the apparent catalytic constant, KMetSO and KTrx the apparent affinity for MetSO and Trx, respectively, and [MetSO] and [Trx] the initial concentration of MetSO and Trx, respectively.
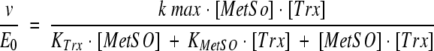 |
(1) |
All kcat, KM, and kcat/KM constants, deduced from fitting the experimental data to a ping-pong model, are summarized in Table 2. With DTT, the kinetic mechanism is also a ping-pong one, as judged by the parallel primary plots (Fig. 2C ▶). Moreover, secondary plots showed a slope cut at the zero ordinate (Fig. 2D ▶). This implies no Michaelis-Menten-type saturation kinetics, and therefore, an infinite value of KDTT and kcat. From the slopes of these plots, it is possible to determine second-order rate constants, which can be considered as kcat/KM values (see Table 2).
Fig. 2.
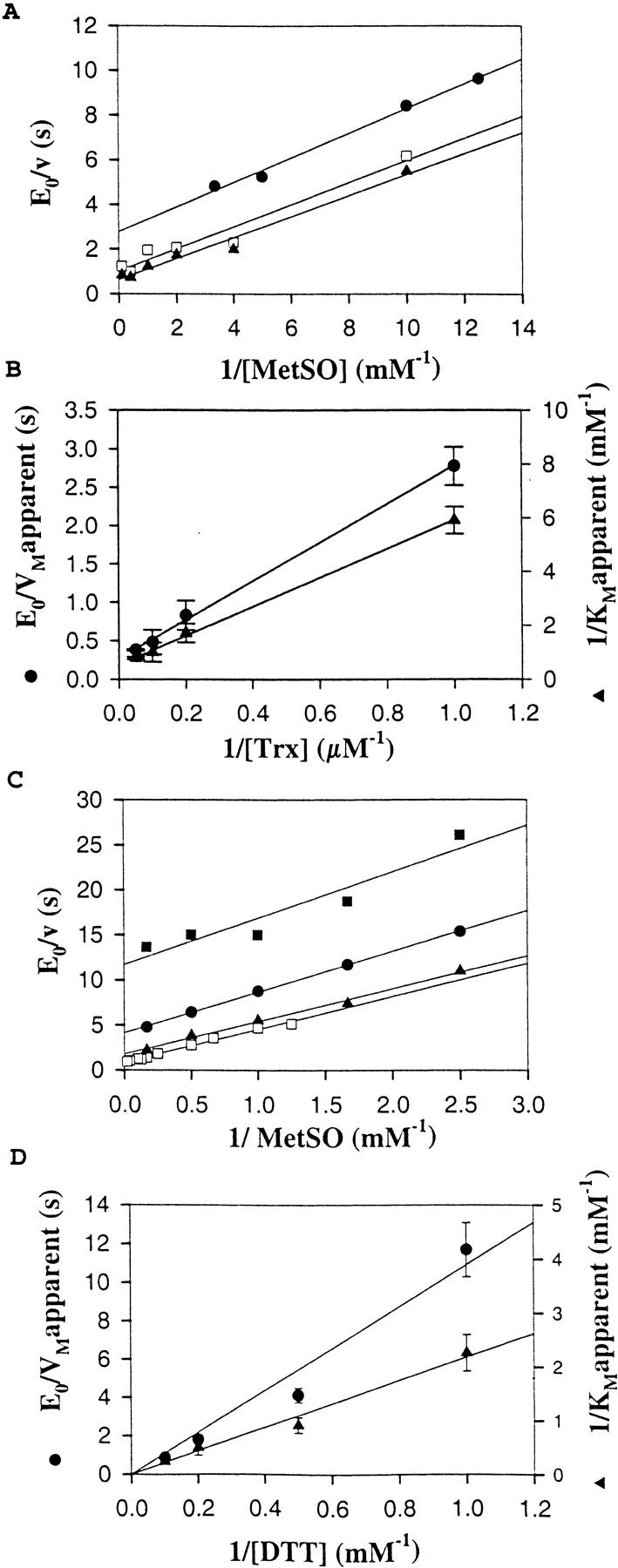
Methionine sulfoxide reductase activity of wild-type MsrA with Trx-regenerating system or DTT. (A) Plots of [E]/v0 (s) versus 1/[MetSO] (mM−1) at [Trx] = 1 μM (filled circles), 5 μM (open squares) and 10 μM (filled triangles). (B) Plots of 1/VMapparent (s) (filled circles) and 1/KMetSO apparent (mM−1) (filled triangles) versus 1/[Trx] (μM−1). (C) Plots of [E]/v0 (s) versus 1/[MetSO] (mM−1) at [DTT] = 1 mM (filled squares), 2 mM (filled circles), 5 mM (filled triangles), and 10 mM (open squares). (D) Plots of 1/VMapparent (s) (filled circles) and 1/KMetSOapparent (mM−1) (filled triangles) versus 1/[DTT] (mM−1).
Table 2.
Kinetic parameters for the methionine sulfoxide reductase activity of wild-type MrsA and truncated mutant MsrAs
| Enzyme (reductant) | kcat (s−1) | KMetSO (mM) | kcat/KMetSO (M−1 · s−1) | KReductant (μM) | kcat/KReductant (M−1 · s−1) |
| Wild-type (Trx) | 3.7 ± 0.5 | 1.9 ± 0.2 | 2020 ± 500 | 10 ± 2 | (4.0 ± 0.1) · 105 |
| Wild-type (DTT) | Infinite | Not determinable | 280 ± 20a | Infinite | 83 ± 7a |
| N-truncated (Trx) | 3.7 ± 0.2 | 8.1 ± 0.8 | 320 ± 50 | 16 ± 3 | (1.5 ± 0.3) · 105 |
| C-truncated (DTT) | Infinite | Not determinable | 290 ± 40a | Infinite | 120 ± 22a |
| N + C-truncated (DTT) | Infinite | Not determinable | 16.2 ± 0.9a | Infinite | 43 ± 18a |
Reactions were carried out in Tris-HCl 50 mM, EDTA 2 mM buffer, pH 8 at 25°C as described in Materials and Methods. Data presented were obtained by fitting the experimental data to a ping-pong mechanism.
a These values were deduced from the slopes of the secondary plots and can be considered as equivalent to a kcat/KM constant.
This mechanism can be described by the following equations, where R represents the reductant (Trx or DTT) and Eox the sulfenic acid intermediate:
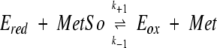 |
(2) |
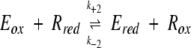 |
(3) |
In this case, the reciprocal rate constant kcat/KMetSO for the net forward reaction of the reduced enzyme with MetSO is defined as k+1 − k−1, and may be regarded as k+1 because the partial reaction shown in Equation 2 is irreversible. Equation 3 can be considered in first approximation as irreversible. Thus, the reciprocal kcat/KTrx for the regeneration step may be regarded as k+2. The fact that the kcat/KMetSO values are not significantly different for Trx and DTT showed that the reductant does not affect the reaction of the reduced enzyme with MetSO (Eq. 2), as expected for a ping-pong mechanism. The lack of enzyme saturation observed with DTT as reductant can be explained as follows: either the rate of formation of enzyme–DTT complexes is slower than the rate of enzyme reduction within the complexes or no specific enzyme–DTT complexes are formed at all. In the latter case, DTT can be considered as a second-order reaction probe. Comparison of the second-order rate constant kcat/KReductant for the regeneration of the reduced enzyme (Eq. 3) showed a higher efficiency of Trx compared to DTT by a factor of 4 × 103. The high kcat/KReductant value with Trx and the fact that saturation kinetics were observed with a low KReductant classify Trx as a specific substrate of MsrA. This is in accord with data that showed that growth of a Met− E. coli strain on MetSO requires the reduction of MsrA by thioredoxin (Russel and Model 1986; Lin 1999).
As expected, truncated enzymes also presented a "ping-pong" kinetic mechanism (data not shown). Kinetic parameters of N-truncated MsrA in the presence of the Trx-regenerating system were obtained by using the "ping-pong" equation described in the Methods and Materials section (see Table 2). The KMetSO was about fourfold higher for N-truncated MsrA than for the wild type, with a same kcat of 3.7 sec−1, whereas no significant effect was observed for the Trx apparent affinity.
As for the wild type, no saturation kinetics were observed for the C-truncated and the double-truncated MsrAs with DTT at concentrations up to 300 mM. For the C-truncated mutant, the kcat/KM values for MetSO and DTT determined from the plot of the rate against the substrate concentration were not significantly different from those of the wild type (see Table 2). For the double-truncated mutant, the efficiency of the sulfenic acid intermediate reduction by DTT was not affected (kcat/KDTT value of 43 M−1 · sec−1 compared to 83 M−1 · sec−1 for the wild-type enzyme), the efficiency of MetSO reduction was decreased 17-fold.
Discussion
In a previous study carried out on the E. coli MsrA, it was shown that in the absence of reductant, 2 moles of methionine were formed per mole of enzyme for the wild type, whereas only 1 mole was formed for mutants in which either Cys-198 or Cys-206 or both were mutated. Concomitantly to the methionine release, formation of a sulfenic acid on Cys-51 was also shown. Together, the results supported a reductase activity not dependent on the presence of Cys-198 and Cys-206. This is also supported by inspection of the structures of MsrAs from E. coli and Bos taurus, which have been recently described (Lowther et al. 2000b; Tête-Favier et al. 2000). Indeed, both structures show a central core, which contains the active site. In the case of E. coli MsrA, the central core is wrapped by two extended chains composed of the N-terminal (amino acids 1–42) and of the C-terminal (amino acids 183–211) ends, which in the latter case includes Cys-198 and Cys-206. Moreover, the N-terminal end and the amino acids 193–211 of the C-terminal end are not in a direct contact with the active site. Such positionings suggested that truncated N- or C-terminal end or both should not drastically destabilize the structure of the central core. This is exactly what we observed. All truncated forms can be folded either in vivo or in vitro. This is confirmed by CD spectra that showed no significant difference in α-helix content compared to the wild type (curves not shown). For the C-terminal enzyme truncated after position 194, a stoichiometry of 1 mole of methionine per mole of truncated enzyme was determined. This result confirms no implication of Cys-198 and Cys-206 in the reduction of MetSO. Moreover, the absence of recycling activity in the presence of Trx definitively proves that the sulfenic intermediate cannot be reduced by Trx. The reductase activity observed for all truncated forms is also an indication that no gross structural alterations were provoked within the active site by the truncations. However, this did not exclude significant effects on the catalytic efficiency. To evaluate such possible effects, the kinetic mechanism was determined and the catalytic constants and the affinities for both methionine sulfoxide and Trx or DTT of MsrA forms were compared. Clearly, the experimental data obtained on the wild-type fit a ping-pong kinetic mechanism, whatever the reductant. However, for the reductants, kinetic saturation was only observed with Trx with an apparent high affinity. This suggests specific interactions between Trx and MsrA at least under the latter's oxidized Cys-198/Cys-206 disulfide state. The fact that DTT is likely not recognized by MsrA can be one of the factors responsible for the low reduction efficiency for DTT. The catalytic efficiency of the wild type for MetSO reduction (kcat/KMetSO) is, however, at least 100-fold less efficient compared with other enzymatic systems involving sulfenic acid intermediates like peroxidases (Parsonage et al. 1993, Crane et al. 2000). In contrast, the catalytic efficiency for Trx is high, and similar to that described for other systems (Lennon and Williams 1997; Zhong and Holmgren 2000). This is mainly due to a KM effect (high KM for MetSO and low KM for Trx) and not to a kcat effect.
As expected, truncation at the N- or C-terminal end, or both, does not change the efficiency of the reduction of the sulfenic acid intermediate. In contrast, the efficiency of the sulfenic acid intermediate formation, kcat/KMetSO, is significantly decreased by the double truncation. An explanation is that the double truncation can provoke slight conformational changes of the active site and thus decreases the affinity for MetSO.
The present and previous studies from our group have clearly shown that MsrA from E. coli bearing only one cysteine at position 51 possesses a methionine sulfoxide reductase activity in the absence of reductant (Boschi-Muller et al. 2000). It was also shown that the regeneration of oxidized Cys-51 requires the intervention of Cys-198 and Cys-206 and of the Trx/Trx reductase recycling system while the fourth cysteine at position 86 is not important for catalysis. Inspection of alignment of the protein sequences of MsrAs whose activity has been characterized either biochemically or by gene inactivation suggests different subclasses (Fig. 1 ▶). The first one contains the three essential Cys-51, 198, and 206, and is well represented by E. coli and bovine MsrAs. The second subclass possesses only the essential Cys-51 and Cys-198. The third one has no Cys-198 and Cys-206, but has a Cys at position 54. All of them are active with Trx. This thus implies that the second and the third subclasses of MsrAs use different means to regenerate Cys-51. For the second subclass, an extension at the N- or C-terminal ends or at both is observed except for Saccharomyces cerevisiae. For MsrAs having a C-terminal extension as in Streptococcus pneumoniae and Neisseria gonnhorroeae, two conserved ms containing a cysteine, i.e., GGLRYCIN and SGCGWPSF can be ideied when aligned with putative sequences corresponding to the C-terminal extension. One of these two cysteines can be involved in the reduction of the Cys-51/Cys-198 disulfide bond unless Trx directly reduces this disulfide bond. In the latter case, this would imply accessibility or/and reactivity of the Cys-51/Cys-198 disulfide bond towards Trx different from that of E. coli MsrA. Moreover, N. gonnhorroeae MsrA also contains an N-terminal extension with two cysteines located in a Trx-like signature. This signature can also be involved in the regeneration of Cys-51. The situation in S. cerevisiae MsrA is different. No extension is observed. Apart from Cys-51 and Cys-198, three other cysteines at position 49, 68, and 86 are present. As expected from studies carried out on the E. coli enzyme, Cys-86 should have no role in catalysis. The other residues 49 and 68 in the E. coli enzyme are located in a loop between the β1A-strand and the α1-helix and at the beginning of the β2-strand, respectively. Only the distance between residues 49 and 51 seems to be compatible with a role of Cys-49 in the regeneration of Cys-51 for the S. cerevisiae enzyme. The third subclass represented by the Bacillus subtilis enzyme has a Trx-like signature C51FWC54. A likely mechanism for regeneration of Cys-51 is the attack of the sulfenic acid intermediate by Cys-54 followed by reduction of the Cys-51/Cys-54 disulfide bond by Trx. Finally, alignment of putative MsrAs (not yet characterized) suggests that a new subclass of MsrA represented by the Rhodobacter capsulatus sequence can exist, which only bears a Cys-51. If the latter MsrA is active in vivo, four alternative mechanisms in Cys-51 regeneration would be at least operative. Studies are underway to validate these alternative mechanisms.
Materials and methods
Production and purification of wild-type and mutant E. coli MsrAs
Plasmid pETMsrA was constructed by cloning the E. coli msrA ORF into the plasmid pET24c in the NdeI and the SacI sites. Truncation of the N-terminal part of E. coli MsrA was obtained by deletion of the 123 bp of the msrA coding sequence. This deletion was obtained by NdeI digestion of a mutated plasmid containing a second NdeI site between the 121st and the 126th bp. Truncation of the C-terminal part was obtained by introducing by site-directed mutagenesis a stop codon (TAA) in place of the Tyr195 codon. Site-directed mutageneses were performed using the Quickchange site-directed mutagenesis kit (Stratagene).
The E. coli strain used for MsrA production was BL21(DE3)pLysS transformed with a pETMsrA plasmid (plasmid containing the wild-type or mutant msrA gene under the control of the T7 promoter). The overexpression of MsrAs was performed by the addition of 1 mM IPTG in the culture medium at 0.6 A600. After 3 h of induction, cultures were harvested by centrifugation and resuspended in buffer A (50 mM Tris-HCl, 2 mM EDTA, pH 8) containing 20 mM dithiothreitol (DTT).
Purification of wild-type and C-truncated MsrAs was performed as previously described by Boschi-Muller et al. (2000).
For purification of both N-truncated MsrAs, pellets obtained after sonication were resuspended in buffer A containing urea 6 M. The contaminating proteins and nucleic acids were removed by applying the enzymatic solution onto a Q-Sepharose column equilibrated with buffer A containing urea 2 M using a fast protein liquid chromatography system (Amersham Pharmacia Biotech). Elution was performed in two steps: first by a linear gradient of urea (2–0 M), and secondly by a linear gradient of KCl (0–0.4 M). N-truncated MsrAs were eluted at 250 mM KCl. At this stage, enzymes were pure as checked by electrophoresis on 12.5 % SDS-polyacrylamide gel (Laemmli 1970), followed by Coomassie Brilliant Blue R-250 staining.
Purified enzymes were stored at −20°C in the presence of 20 mM DTT and 45% ammonium sulfate. Under these conditions, the enzymes were stable for several weeks. Their molar concentrations were determined spectrophotometrically, using a molar absorptivity at 280 nm of 34632 M−1 cm−1, deduced from the method of Scopes (Scopes 1974), for wild-type and N-truncated mutant MsrAs, and of 28944 M−1 cm−1 for C-truncated and double truncated mutant MsrAs.
Quaication of the free cysteine content with 5,5′-dithiobis(2-nitrobenzoate)
Cysteine content of protein samples was determined using DTNB under nondenaturing and denaturing conditions, either after incubation or without incubation with 10 mM MetSO but without the addition of any exogenous reducing system. Progress curves of 3-carboxy-4-nitrobenzenethiol (TNB−) production for wild-type and mutant enzymes were recorded at 412 nm in buffer A for nondenaturing conditions. For denaturing conditions, SDS was added to a final concentration of 10%, and the enzyme solution was heated for 10 min at 70°C. Enzyme concentrations were 7.35 and 14.7 μM, and DTNB concentration was 300 μM. The amount of TNB− formed was calculated using an extinction coefficient at 412 nm of 13,600 M−1 cm−1.
Enzyme kinetics
The ability of wild-type and mutant MsrAs to reduce free MetSO was assayed by using D,L-MetSO as a substrate and either 10 mM DTT or a Trx-regenerating system (100 μM Chlamydomonas reinhardtii Trx, 1.2 μM Arabidopsis thaliana Trx reductase, and 0.3 mM NADPH) as previously described by Boschi-Muller et al. (2000).
Initial rate data were fitted to a ping-pong model with the program Sigmaplot (Jandel Scieic Software) as follows:
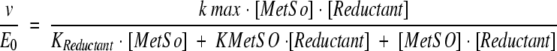 |
Stoichiometry of Met formation in the absence of regenerating system
The reaction mixture containing 10 mM MetSO and 100–500 μM of enzyme was incubated at 25°C for 10 min in buffer A. Then, the Met formed was quaied as previously described by Boschi-Muller et al. (2000).
Acknowledgments
This research was supported by the "Centre National de la Recherche Scieique" (CNRS), the program "Physique et Chimie du Vivant 2000" of the CNRS, the University Henry Poincaré, Nancy I, and the Association pour la Recherche sur le Cancer (ARC-No 5436). We are very grateful to A. Orly for his technical help, to S. Sanglier-Cianferani and Dr A. Van Dorsselear for determining the molecular weights of proteins, and to Dr. J.P. Jacquot for his help in production and purification of C.reinhardtii Trx and A thaliana Trx reductase.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
DTNB, 5,5′-dithiobis(2-nitrobenzoate)
DTT, dithiothreitol
KDTT, KM for DTT
KMetSO, KM for MetSO
MetSO, methionine sulfoxide
MsrA, methionine sulfoxide reductase
TFA, trifluoroacetic acid
TNB−, 3-carboxy-4-nitrobenzenethiol
Trx, thioredoxin
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.10701.
References
- Abrams, W.R., Weinbaum, G., Weissbach, L., Weissbach, H., and Brot, N. 1981. Enzymatic reduction of oxidized alpha-1-proteinase inhibitor restores biological activity. Proc. Natl. Acad. Sci. 78 7483–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschi-Muller, S., Azza, S., Sanglier-Cianferani, S., Talfournier, F., Van Dorsselear, A., and Branlant, G. 2000. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J. Biol. Chem. 275 35908–35913. [DOI] [PubMed] [Google Scholar]
- Crane, E.J., Yeh, J.I., Luba, J., and Claiborne, A. 2000. Analysis of the kinetic and redox properties of the NADH peroxidase R303M mutant: Correlation with the crystal structure. Biochemistry 39 10353–10364. [DOI] [PubMed] [Google Scholar]
- El Hassouni, M., Chambost, J.P., Expert, D., Van Gijsegem, F., and Barras, F. 1999. The minimal gene set member msrA, encoding peptide methionine sulfoxide reductase, is a virulence determinant of the plant pathogen Erwinia chrysanthemi. Proc. Natl. Acad. Sci. 96 887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, C.S., Illades-Aguiar, B., Casillas-Martinez, L., and Setlow, P. 1998. In vitro and in vivo oxidation of methionine residues in small, acid-soluble spore proteins from Bacillus species. J. Bacteriol. 180 2694–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschel, L., Hansel, A., Schonherr, R., Weissbach, H., Brot, N., Hoshi, T., and Heinemann, S.H. 1999. Molecular cloning and functional expression of a human peptide methionine sulfoxide reductase (hMsrA). FEBS Lett. 456 17–21. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–687. [DOI] [PubMed] [Google Scholar]
- Lennon, B.W. and Williams, C.H. 1997. Reductive half-reaction of thioredoxin reductase from Escherichia coli. Biochemistry 36 9464–9477. [DOI] [PubMed] [Google Scholar]
- Lin, T.Y. 1999. G33D mutant thioredoxin primarily affects the kinetics of reaction with thioredoxin reductase. Probing the structure of the mutant protein. Biochemistry 38 15508–15513. [DOI] [PubMed] [Google Scholar]
- Lowther, W.T., Brot, N., Weissbach, H., Honek, J.F., and Matthews, B.W. 2000a. Thiol–disulfide exchange is involved in the catalytic mechanism of peptide methionine sulfoxide reductase. Proc. Natl. Acad. Sci. 97 6463–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther, W.T., Brot, N., Weissbach, H., and Matthews, B.W. 2000b. Structure and mechanism of peptide methionine sulfoxide reductase, an "anti-oxidation" enzyme. Biochemistry 39 13307–13312. [DOI] [PubMed] [Google Scholar]
- Moskovitz, J., Berlett, B.S., Poston, J.M., and Stadtman, E.R. 1997. The yeast peptide-methionine sulfoxide reductase functions as an antioxidant in vivo. Proc. Natl. Acad. Sci. 94 9585–9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz, J., Flescher, E., Berlett, B.S., Azare, J., Poston, J.M., and Stadtman, E.R. 1998. Overexpression of peptide-methionine sulfoxide reductase in Saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proc. Natl. Acad. Sci. 95 14071–14075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz, J., Jenkins, N.A., Gilbert, D.J., Copeland, N.G., Jursky, F., Weissbach, H., and Brot, N. 1996. Chromosomal localization of the mammalian peptide-methionine sulfoxide reductase gene and its differential expression in various tissues. Proc. Natl. Acad. Sci. 93 3205–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz, J., Rahman, M.A., Strassman, .J., Yancey, S.O., Kushner, S.R., Brot, N., and Weissbach, H. 1995. Escherichia coli peptide methionine sulfoxide reductase gene: Regulation of expression and role in protecting against oxidative damage. J. Bacteriol. 177 502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsonage, D., Miller, H., Ross, R.P., and Claiborne, A. 1993. Purification and analysis of streptococcal NADH peroxidase expressed in Escherichia coli. J. Biol. Chem. 15 3161–3167. [PubMed] [Google Scholar]
- Russel, M. and Model, P. 1986. The role of thioredoxin in filamentous phage assembly. Construction, isolation, and characterization of mutant thioredoxins. J. Biol. Chem. 261 14997–15005. [PubMed] [Google Scholar]
- Sadanandom, A., Piffanelli, P., Knott, T., Robinson, C., Sharpe, A., Lydiate, D., and Murphy, D. 1996. Ideication of a peptide methionine sulphoxide reductase gene in an oleosin promoter from Brassica napus. Plant J. 10 235–242. [DOI] [PubMed] [Google Scholar]
- Scopes, R.K. 1974. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 59 277–282. [DOI] [PubMed] [Google Scholar]
- Sun, H., Gao, J., Ferrington, D.A., Biesada, H., Williams, T.D., and Squier, T.C. 1999. Repair of oxidized calmodulin by methionine sulfoxide reductase restores ability to activate the plasma membrane Ca-ATPase. Biochemistry 38 105–112. [DOI] [PubMed] [Google Scholar]
- Tête-Favier, F., Cobessi, D., Boschi-Muller, S., Azza, S., Branlant, G., and Aubry, A. 2000. Crystal structure of the Escherichia coli peptide methionine sulphoxide reductase at 1.9 Å resolution. Structure 8 1167–1178. [DOI] [PubMed] [Google Scholar]
- Wizemann, T.M., Moskovitz, J., Pearce, B.J., Cundell, D., Arvidson, C.G., So, M., Weissbach, H., Brot, N., and Masure, H.R. 1996. Peptide methionine sulfoxide reductase contributes to the maintenance of adhesins in three major pathogens. Proc. Natl. Acad. Sci. 93 7985–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, L. and Holmgren, A. 2000. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 275 18121–18128. [DOI] [PubMed] [Google Scholar]