

Abstract
To assess the role of quaternary stability on the properties of Escherichia coli phosphofructokinase (PFK), a disulfide bond has been introduced across the subunit interface containing the allosteric binding sites in E. coli phosphofructokinase by changing N288 to cysteine. N288 is located in close proximity to the equivalent residue on an adjacent subunit. Although SDS-PAGE of oxidized N288C indicates monomeric protein, blocking the six native cysteine residues with N-ethyl maleimide (NEM) reveals dimers of N288C on non-native gels. Subsequent addition of dithiothreitol (DTT) to NEM-labeled N288C regenerates the monomer on SDS-PAGE, reflecting the reversibility of intersubunit disulfide bond formation. KSCN-induced hybrid formation between N288C and the charged-tagged mutant E195,199K exhibits full monomer–monomer exchange only upon DTT addition, providing a novel assessment of disulfide bond formation without NEM treatment. N288C also exhibits a diminished tendency toward nonspecific aggregation under denaturing conditions, a phenomenon associated with monomer formation in PFK. Pressure-induced dissociation and urea denaturation studies further indicate that oxidized N288C exhibits increased quaternary stability along both interfaces of the tetramer, suggesting a synergistic relationship between active site and allosteric site formation. Although the apparent binding affinities of substrates and effectors change somewhat upon disulfide formation in N288C, little difference is evident between the maximally inhibited and activated forms of the enzyme in oxidizing versus reducing conditions. Allosteric influence, therefore, is not correlated to subunit–subunit affinity, and does not involve substantial interfacial rearrangement.
Keywords: Phosphofructokinase, disulfide cross-link, allosteric regulation, subunit interactions, pressure dissociation, urea denaturation, hybrid formation
Phosphofructokinase from E. coli is a tetrameric protein with subunits oriented with two distinct dimer–dimer interfaces (Shirakihara and Evans 1988). The four active sites are positioned along one interface, and bind the substrate fructose-6-phosphate (Fru-6-P) cooperatively. The four allosteric sites are situated along the other dimer–dimer interface to which either the activator MgADP or the inhibitor phospho(enol)pyruvate (PEP) binds. Both allosteric ligands act by altering the affinity of the enzyme for Fru-6-P. Dissociation studies using the denaturants guanidine hydrochloride (Teschner and Garel 1989), KSCN (Deville-Bonne et al. 1989), and urea (Le Bras et al. 1989) suggest that there is a specific order to the formation and loss of quaternary structure in E. coli PFK corresponding to the pathway:
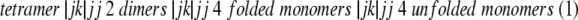 |
(1) |
Each ligand stabilizes the interface to which it binds such that Fru-6-P maintains the catalytically active tetramer against dissociation to inactive dimer, and MgADP and PEP bind to, and stabilize, the dimer against monomer formation (Deville-Bonne et al. 1989; Le Bras et al. 1989).
Asparagine, at position 288, has been replaced with cysteine via site-directed mutagenesis to encourage the formation of two disulfide bonds along the dimer–dimer interface to which the allosteric ligands bind. Asparagine 288 was chosen because it is very close to its symmetry-related equivalent across the interface, within a loosely ordered loop between β-strands, and removed from direct interactions with ligand binding sites (Shirakihara and Evans 1988). It seemed likely that upon oxidation these proximate cysteine residues would result in the formation of a single intersubunit disulfide bridge per dimer in the N288C variant. There also should be no interference in disulfide bond formation by sulfhydryl groups from any of the six native cysteine residues per subunit, because there are no native cysteine residues closer than 16 Å to N288 across the effector-site interface (Shirakihara and Evans 1988). Urea denaturation, pressure dissociation, and KSCN-induced subunit exchange confirm that not only is the dimer stabilized in oxidized N288C, but also that the tetramer/dimer equilibrium is likewise displaced in favor of active tetramer.
We have investigated the impact on allosteric response of imposing this stabilization on the active tetramer of E. coli PFK using a thermodynamic linked-function approach previously established (Weber 1972Weber 1975; Reinhart 1983 Reinhart 1985 Reinhart 1988; Johnson and Reinhart 1992 Johnson and Reinhart 1994a, 1997). In addition, it has been observed that this cross-linked variant of PFK, when oxidized, is less prone to aggregate to insoluble species in both chemical and pressure dissociation studies, supporting previous suggestions that such nonspecific aggregation is due to incorrect interactions between partially folded monomers (Le Bras et al. 1989; Teschner and Garel 1989).
Results
SDS-PAGE analyses of disulfide bonds in N288C
To establish whether a cysteine introduced at position 288 could form an intersubunit disulfide bridge in an oxidizing environment, SDS-PAGE was performed on wild-type and N288C in the presence and absence of DTT. All samples were denatured prior to loading onto 10% polyacrylamide gels by incubation for 5 min at 100°C in loading buffer containing 100 mM Tris-HCl (pH 6.9), 8% SDS, 25% glycerol, and 0.02 mg/mL bromophenol blue. Initially, utilizing this denaturation procedure, no differences in SDS-PAGE mobilities were detected, with all four samples (wild-type +/− DTT and N288C +/− DTT) behaving as a monomer (data not shown). However, when samples were preincubated for 12 h in 1 mM NEM (a thiol-reactive alkylating agent) prior to their thermal denaturation with SDS, dimers of oxidized N288C were apparent on a denaturing gel, whereas wild-type PFK still runs as a monomer (Fig. 1 ▶). These data suggest that disulfide bonds link monomers together in N288C, and that NEM treatment prevents intrasubunit disulfide exchange from reducing the covalent bond between the engineered cysteine residues once the protein unfolds. (Note that wild-type PFK contains six reduced cysteine residues per subunit.) NEM-labeled N288C migrates as a monomer in SDS-PAGE when DTT is subsequently added, demonstrating the reversible reduction of the disulfide bond in the modified protein (Fig. 1 ▶).
Fig. 1.

SDS-PAGE analysis of NEM-labeled PFK. All PFK samples were reacted with NEM under nondenaturing conditions prior to denaturation at 100°C in the presence of 8% w/v SDS and loaded onto a 10% resolving gel. Molecular weight markers are in lane 1, with their approximate sizes shown to the far left. Lanes 2 and 3 portray NEM-modified wild-type PFK with 0.28 M DTT (lane 2) and no DTT (lane 3) included in the loading buffer. Lanes 4 and 5 contain NEM-modified N288C with 0.28 M DTT (lane 4) and no DTT (lane 5) included in the loading buffer.
Subunit exchange studies
The ability to detect and idey hybrids resulting from subunit exchange between charge-altered and wild-type subunits (see Materials and Methods) was used to verify the presence of an intersubunit disulfide in N288C under oxidizing conditions in the absence of NEM treatment. The charge-tag mutant E195,199K was constructed in which two negatively charged, surface residues, glutamate 195 and 199, were converted, via site-directed mutagenesis, into the positively charged amino acid lysine. The surface residues were selected because they were far removed from ligand binding sites and subunit interfaces, so as not to interfere with the allosteric response of the protein, but were outwardly positioned to confer an altered chromatographic response and gel mobility versus unmodified protein. When mixed with PFK exhibiting a native charge density (wild-type or N288C), the degree to which each PFK is free to equilibrate between tetramer, dimer, and monomer can be assessed by the degree to which hybrid quaternary states with individually varying charge density accumulate.
In Figure 2 ▶, we present the result of native gel electrophoresis on mixtures of charge-tagged PFK and either wild-type (lanes 4,5) or N288C (lanes 6,7) subjected to the same hybridization protocol except that DTT was included in the samples run in lanes 5 and 7. The presence of five independent bands in lane 4, corresponding to the five possible hybrid combinations (4:0, 1:3, 2:2, 3:1, 0:4) upon full subunit exchange, indicates that there are no intersubunit disulfide bonds in native PFK. The addition of DTT to this sample (lane 5) had no impact on results. In contrast, hybrid formation between N288C and E195,199K was restricted to pure dimer exchange under oxidizing conditions (lane 6), whereas the addition of DTT to the sample resulted in complete exchange (lane 7). These data confirm that N288C, unmodified by NEM, possesses disulfide bonds in its quaternary structure that prevents free monomers from forming and exchanging in the presence of KSCN.
Fig. 2.

Native-PAGE analysis of hybrid tetramer formation in E. coli PFK. Lanes 1, 2, and 3 represent pure tetramers of wild-type PFK, N288C, and E195,199K, respectively. Lanes 4–7 are representative of hybrid tetramers formed upon KSCN-induced subunit exchange between wild-type PFK and E195,199K in the absence of DTT (lane 4) and in the presence of 10 mM DTT (lane 5) and between N288C and E195,199K in the absence of DTT (lane 6) and in the presence of 10 mM DTT (lane 7).
Pressure-induced dissociation of N288C
The influence of the putative disulfide bond in N288C on the quaternary association equilibria is further demonstrated by the response of N288C to pressure. Deville-Bonne and Else (1991) have reported that unliganded E. coli PFK begins to dissociate when hydrostatic pressures exceed 800 bar, and we have shown that this dissociation is reversible, and enzymatic activity is recoverable, upon depressurization provided that pressure never exceeds 1200 bar (Johnson and Reinhart 1996). At pressures greater than 1200 bar an irreversible drop in fluorescence intensity of the single tryptophan residue (W311), an increase in W311 accessibility to iodide quenching, and visible accumulation of protein precipitate are evident, suggesting the formation and resulting aggregation of monomeric wild-type PFK in an oxidizing environment at high pressure (data not shown). The variant N288C exhibits similar changes in fluorescence intensity when subjected to pressure in the presence of DTT (Fig. 3 ▶)—the steady-state intensity gradually increases from atmospheric pressure up to 1700 bar, likely due to compaction of the subunits themselves (Heremans and Smeller 1998) and then precipitously and irreversibly drops to roughly 30% of its highest value at 3000 bar due to subunit dissociation. In the absence of DTT, however, the response of N288C to pressure changes markedly. The hydrostatic pressure can be raised to a value of approximately 2300 bar before any decrease in the steady-state fluorescence intensity of oxidized N288C is observed, with roughly 85% of the highest intensity still maintained at 3000 bar. Whereas aggregation can be observed in the wild-type and reduced N288C following depressurization, none is visible in samples of oxidized N288C at identical subunit concentrations. When the maximum pressure subjected on each sample is restricted to 2400, the relative enzymatic activity remaining at least 12 h following depressurization is roughly 6, 50, and 100% for oxidized wild-type PFK, reduced N288C, and oxidized N288C, respectively. Interestingly, up to 90% activity can be recovered from 2400 bar pressurization for wild-type PFK if DTT is included, suggesting that improper interactions between thiol groups native to PFK might partially be responsible for observed monomer aggregation under oxidizing conditions.
Fig. 3.

Hydrostatic pressure dissociation of PFK. Changes in the steady-state intrinsic fluorescence of wild-type PFK (filled circles, open circles) in the absence of DTT and of N288C in the absence (filled squares, open squares) and in the presence of 2 mM DTT (filled triangles, open triangles) as a function of increasing (closed symbols) and decreasing (open symbols) pressure. Samples were allowed to equilibrate at each pressure for 15 min prior to each measurement. Fluorescence intensity remaining after complete depressurization of each sample was constant for at least 12 h.
Urea-induced unfolding of N288C
Enzymatic activity and intrinsic fluorescence intensity of N288C and wild-type PFK were monitored as a function of urea concentration under oxidizing and reducing conditions. Activity changes reflect the dissociation of the tetramer, because the binding of the substrate Fru-6-P requires intact tetramer (Shirakihara and Evans 1988). The formation of monomers results in solvent exposure of W311, located along the regulatory interface, and leads to an approximately 70% decrease in steady-state fluorescence emission (Teschner and Garel 1989). The midpoints of the tetramer-to-dimer and dimer-to-monomer transitions for wild-type PFK remain unaltered by the presence of the DTT (Fig. 4A ▶), whereas the unfolding transitions of N288C differ significantly upon DTT addition (Fig. 4B ▶). In the presence of DTT, denaturation profiles of N288C roughly correspond with those of native PFK, whereas when DTT is not included in the incubation mixture the midpoints of transition in activity and fluorescence are increased by roughly 1.5 and 2 M in urea concentration, respectively. The result of overall tetramer stability by cross-linking the stronger dimer–dimer interface indicates a substantial synergism in the stability of both interfaces.
Fig. 4.

Urea denaturation profiles of E. coli PFK. The relative intrinsic fluorescence (λex = 300 nm) in the absence (filled circles) and presence of 10 mM DTT (open circles), and the relative enzymatic activity in the absence (filled triangles) and presence of 10 mM DTT (open triangles) are presented as a function of urea concentration for wild-type PFK (A) and N288C (B).
Allosteric couplings in N288C
To compare the allosteric response of N288C with intersubunit disulfide bonds along the regulatory interface versus that of the reduced enzyme, the K0.5 for Fru-6-P was monitored as a function of MgADP and PEP concentration in the presence and absence of 2 mM DTT. The Michaelis constant for MgATP is, within error, identical between N288C and wild-type PFK under all solution conditions examined (Km = 40–50 μM). MgATP was kept saturating at 4 mM throughout these studies. Values of K0.5 for Fru-6-P, previously shown to bind in rapid equilibrium (Johnson and Reinhart 1997), were measured at MgADP concentrations ranging from 0 to 1 mM and PEP concentrations ranging from 0 to 60 mM (Fig. 5 ▶). The resulting data were fit to equation 3 to provide the allosteric parameters listed in Table 1.
Fig. 5.

Variation of K0.5 for Fru-6-P with allosteric effector concentration. K0.5 values for Fru-6-P were determined kinetically in the presence of 4 mM MgATP in the absence (open symbols) or presence (closed symbols) of 2 mM DTT. Data were collected at 25°C as a function of either PEP (filled circles, open circles) or MgADP (filled triangles, open triangles) concentrations. Solid lines (2 mM DTT) and dashed lines (0 mM DTT) represent the best fit of these data to equation 3. Values for all parameters deriving from these fits are presented in Table 1.
Table 1.
Ligand dissociation constants from E. coli PFKa
| Protein | Ligand | Other saturating ligands | Designation | Kd (μM) |
| Wild typeb,c | Fru-6-P | — | Kia/b | 420 ± 20 |
| Wild type | Fru-6-P | MgADP | Kia/bx | 23 ± 1 |
| Wild type | Fru-6-P | PEP | Kia/by | (2.6 ± 0.3) × 104 |
| Wild type | MgADP | — | Kix/b | 13 ± 3 |
| Wild type | MgADP | Fru-6-P | Kix/ab | 10 ± 1 |
| Wild type | PEP | — | Kiy/b | 240 ± 20 |
| Wild type | PEP | Fru-6-P | Kiy/ab | (1.5 ± 0.2) × 104 |
| N288Cc | Fru-6-P | — | Kia/b | 161 ± 2 |
| N288C | Fru-6-P | MgADP | Kia/bx | 25 ± 2 |
| N288C | Fru-6-P | PEP | Kia/by | (2.7 ± 0.4) × 104 |
| N288C | MgADP | — | Kix/b | 33 ± 2 |
| N288C | MgADP | Fru-6-P | Kix/ab | 3.0 ± 0.7 |
| N288C | PEP | — | Kiy/b | 200 ± 4 |
| N288C | PEP | Fru-6-P | Kiy/ab | (3.2 ± 0.1) × 104 |
| N288Cd | Fru-6-P | — | Kia/b | 373 ± 2 |
| N288C | Fru-6-P | MgADP | Kia/bx | 28 ± 3 |
| N288C | Fru-6-P | PEP | Kia/by | (3.1 ± 0.4) × 104 |
| N288C | MgADP | — | Kix/b | 130 ± 7 |
| N288C | MgADP | Fru-6-P | Kix/ab | 9.0 ± 0.5 |
| N288C | PEP | — | Kiy/b | 480 ± 10 |
| N288C | PEP | Fru-6-P | Kiy/ab | (3.6 ± 0.2) × 104 |
a Kinetic data were collected at 25°C with MgATP saturating at 4 mM.
b As reported by Johnson and Reinhart (1994a, 1997).
c Dissociation constants determined in the presence of 2 mM DTT.
d Dissociation constants determined in the absence of DTT.
The magnitude of the total inhibition of Fru-6-P affinity by PEP is somewhat greater when cystine has been reduced to free thiols (Qay/b = 0.006 ± 0.002) than when intersubunit disulfide bonds are intact (Qay/b = 0.013 ± 0.001) or in wild-type PFK (Qay/b = 0.016 ± 0.003) (Johnson and Reinhart 1997). In contrast, MgADP exerts a slightly greater enhancing influence on Fru-6-P binding within both oxidized N288C (Qax/b = 14.5 ± 0.4) and wild-type PFK (Qax/b = 18.3 ± 1.1) (Johnson and Reinhart 1994a) than in the presence of DTT (Qax/b = 10.9 ± 0.2). These differences in thermodynamic couplings are clearly associated with differences in the affinity of Fru-6-P in the absence of allosteric ligands (see Table 1). Although oxidized N288C is similar to wild-type in its affinity for Fru-6-P in the absence of allosteric effectors (Kia/b), the presence of disulfide bonds along the regulatory interface lowers the protein's affinity for Fru-6-P, MgADP (Kix/b), and PEP (Kiy/b) relative to reduced N288C. However, the affinity of Fru-6-P for N288C in the saturating presence of either allosteric effector does not depend upon the presence of DTT, as values for Kia/bx and Kia/by remain essentially identical to the values exhibited by wild-type PFK. Clearly, stabilizing the quaternary structure via covalent disulfide bonds along the allosteric dimer–dimer interface has essentially no impact on the maximally inhibited or activated state of the protein with respect to allosteric perturbations by PEP and MgADP.
Discussion
Because oxidized N288C only appears as dimers on SDS-PAGE if the sample has been pretreated with NEM, a possible interpretation might be that NEM induces a conformation change that only then allows for the formation of intersubunit cross-links. However, both urea and pressure dissociation profiles of N288C, unaltered by NEM, clearly indicate an enhanced stability relative to wild-type protein that is lost upon the addition of DTT, and subunit exchange studies indicate clear protection for N288C unaltered by NEM against quaternary disruption. NEM itself, therefore, should not be considered a causal agent in the formation of disulfide bonds, but rather its introduction served to prevent the loss of cross-links upon SDS-induced denaturation by blocking native free sulfhydryls against intramolecular disulfide exchange.
The introduction of novel disulfide bonds into globular proteins to increase stability has in many cases been shown to have the opposite effect (Betz 1993). Whereas the free energy of folding is thought to be naturally enhanced by reducing the configurational entropy of the denatured state (Flory 1956), the intrinsic strain accompanying the stringent bond angles and Cα–Cα atomic separation of the disulfide bond often leads to a net destabilization of the protein (Betz 1993). Chemical and pressure dissociation studies here, however, both suggest that the effector-site interface has been substantially stabilized by the presence of a disulfide bridge introduced between cysteines at positions 288 in E. coli PFK. Moreover, there is also an increase in stability conferred on the smaller, active-site interface containing no mutational substitutions. Although the formation of each of the two interfaces within E. coli PFK is generally treated as a distinct, separable process (Deville-Bonne et al. 1989; Le Bras et al. 1989; Teschner and Garel 1989), our data suggests that there is an apparent synergism in their ultimate contributions to full tetramer stability. The apparent stabilization of the noncross-linked active site dimer–dimer interface may be either the result of structural interactions between the two interfaces or a natural consequence of the Mass Action Law as it relates to the dissociation equilibria depicted in Scheme 1.
The potential influence of disulfide bridges on the homotropic and heterotropic interactions across the PFK tetramer is particularly intriguing given that (1) the cooperative binding of Fru-6-P has been shown to vary inversely with the over-all stability of E. coli PFK (Le Bras et al. 1995); (2) the presence of PEP increases the rate of inactivation of the PFK tetramer, presumably by weakening the active site dimer–dimer interface of E. coli PFK (Le Bras et al. 1989); and (3) the regulation of a similar PFK from Thermus thermophilus has been shown to result largely from tetramer–dimer conversion induced by the binding of PEP (Xu et al. 1990). Together, these data suggest that the strengthening of the allosteric-site interface of E. coli PFK might directly result in at least a partial dampening of homotropic cooperativity in Fru-6-P binding and, via the indirect increase in stability of the active-site interface, lessen the degree of inhibition by PEP. Neither potential effect was observed in N288C, however. In activity saturation profiles, the substrate Fru-6-P continued to bind cooperatively with no change in Hill number to both oxidized and reduced N288C when MgATP concentrations were maintained at 4 mM (data not shown). Cooperativity, Fru-6-P decreased as a function of the concentration of both MgADP and PEP from Hill number values near 3.6 in the absence of an effector to near 1.2 in their saturating presence, as is characteristic of wild-type PFK (Blangy et al. 1968). Binding affinities of PFK for each ligand individually are somewhat perturbed by the presence of the disulfide bond. Oxidized N288C exhibits a twofold increase in the dissociation constant of PEP (Kiy/b) and, most notably, a 10-fold increase in that of MgADP (Kix/b) relative to wild-type protein, whereas reduced N288C has affinities for the allosteric effectors more closely matching those of unaltered PFK. However, the magnitude of inhibition by binding PEP (Qay/b) as well as activation by MgADP (Qax/b) are unaffected by the presence of thiol cross-links in N288C relative to wild-type E. coli PFK. The disulfide bond within N288C is, therefore, minimally invasive relative to the allosteric communication in E. coli PFK.
The absence of a noteworthy impact of a sulfhydryl cross-link at position 288 on the allosteric response of bacterial PFK is consistent with structural comparisons between the "activated" and "inhibited" states of PFK from Bacillus stearothermophilus, an enzyme sharing similar structure and 54% amino acid identity with that of E. coli. No significant changes along the larger, allosteric-site interface can be seen when overlapping the crystal structure of PFK bound to Fru-6-P and MgADP (activated state) with that of PFK crystallized in the presence of 2-phosphoglycolate to simulate PEP binding (inhibited state) (Schirmer and Evans 1990). Specifically, the gamma carbon between neighboring N288 residues across the interface is repositioned by less than 0.5 Å when comparing the two crystal structures. However, our data have shown that the presence of the disulfide bond also strengthens the active site subunit interface. The fact that the tightening of this interface does not alter the allosteric properties suggests that the structural shift along the active site interface noted when comparing the two structures just mentioned may not be as central to the allosteric communication as originally proposed (Schirmer and Evans 1990).
Materials and methods
Materials
All chemical reagents used in buffers, PFK purification, and fluorescence and enzymatic assays were of analytical grade, purchased from either Sigma, Fisher, or Aldrich. The Matrex Gel Blue A-agarose resin for affinity chromatography was purchased from Amicon Corporation. Creatine phosphate, creatine kinase, and the sodium salts of Fru-6-P and PEP were obtained from Sigma. The sodium salt of ATP (containing less than 0.5% ADP contamination as determined by enzymatic assay) and the coupling enzymes aldolase, triosephosphate isomerase, and glycerol-3-phosphate dehydrogenase in ammonium sulfate suspensions were obtained from Boehringer Mannheim. Coupling enzymes were dialyzed extensively against a buffer comprised of 50 mM MOPS-KOH, 100 mM KCl, 5 mM MgCl2, and 100 μM EDTA at pH 7.0. Punctilious grade absolute ethanol, obtained from Quantum Chemicals, was used as the pressurizing medium in hydrostatic pressure studies. All enzymes used for site-directed mutagenesis were obtained from either Promega or New England Biolabs, Inc. Deionized distilled water was used throughout.
Site-directed mutagenesis
The plasmid pRZ3 (Zheng and Kemp 1992), which originally contained the gene for E. coli PFK, was a generous gift from Dr. Robert Kemp (University of Health Sciences/The Chicago Medical School, North Chicago, IL). The gene for PFK was removed from pRZ3 by digestion with HindIII and BamHI, and ligated into pAlter1TM previously digested with HindIII and BamHI. The pfk gene promoter region was removed by introducing a HindIII site just upstream of the coding region at position −17 using the protocol for Altered Sites In Vitro Mutagenesis System as provided by Promega, followed by digestion of the ideied mutant plasmid DNA with HindIII and BamHI and recloning of the pfk gene into the HindIII/BamHI sites of pAlter1. The resulting plasmid (pGDR16) contained the gene for PFK in pAlter1 under control of the Lac promoter. pGDR16 was subsequently used to express wild-type PFK and to construct all mutants.
Mutagenesis was performed using the protocol of the Altered Sites II In Vitro Mutagenesis System from Promega. All oligonucleotides were synthesized using an Applied Biosystems 392 DNA/RNA synthesizer at the Gene Technologies Laboratory of the Institute of Developmental and Molecular Biology at Texas A&M University. The sequences of the mutagenic primers for N→C288, E→K195,199, and W→Y311, respectively, were designed to be complementary to the coding strand as follows:
 |
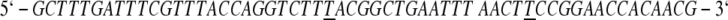 |
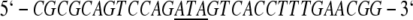 |
(Underlined bases correspond to sites of mutation.) To verify incorporation of the desired mutation, double-stranded DNA samples were prepared for sequencing using a 1/4 reaction scheme with ABI Big-Dye Terminators via the Sanger dideoxy method and sent to the Gene Technologies Laboratory of Texas A&M University for automated gel reading using an ABI Prism 377XL Sequencer.
PFK purification
Individual colonies of E. coli DF1020, from which the pfk-1 and pfk-2 genes have been deleted (Daldel 1983; Hellinga and Evans 1985), carrying the vector of interest were used to inoculate 5 mL of standard Luria-Bertaini (LB) media. The culture was grown overnight in the presence of either 15 μg/mL tetracycline or 100 μg/mL ampicillin, from which 1 mL was transferred to 2 L of LB. Typically, 6 to 8 g of cells per liter were harvested and sonicated in the presence of Buffer A containing 50 mM TRIS-HCl (pH 7.5), 5 mM MgCl2, 2 mM DTT, and 0.1 mM EDTA. Overexpressed PFK was then purified via a modification of the method of Kotlarz and Buc (1982), as described previously (Johnson and Reinhart 1992).
Protein determination
Protein concentrations were determined using the BCA Protein Assay Reagent. In the absence of nucleotides, purified PFK concentrations were verified using absorbance readings [ɛ = 0.6 cm2mg−1) (Kotlarz and Buc 1977) at 278 nm.
Enzyme activity determination
Activity measurements were carried out at 25°C in 1.0 mL of Buffer F (50 mM EPPS-KOH adjusted to pH 8.0 containing 10 mM MgCl2, 10 mM NH4Cl, and 0.1 mM EDTA) containing 0.2 mM NADH, 250 μg of aldolase, 50 μg of glycerol-3-phosphate dehydrogenase, 5 μg of triosephosphate isomerase, 2 mM DTT (when included), 4 mM MgATP, and the indicated concentrations of Fru-6-P, MgADP, and PEP. All nucleotide additions were accompanied by equimolar concentrations of MgCl2, leaving a constant 10 mM Mg+2 in excess. In measurements requiring MgATP regeneration, 1 mM creatine phosphate and 10 μg/mL creatine kinase were also included in the assay mixture. The enzymatic reaction was initiated with 5–10 μL of suitably diluted PFK. The steady-state reaction rate was determined by monitoring the decrease in absorbance at 340 nm with respect to time with a model 640 Beckman UV/VIS spectrophotometer. A unit of activity is defined as the amount of enzyme required to produce 1 μmole of Fru-1,6-BP per minute.
Establishing oxidation/reduction solution conditions
Routine storage of purified proteins in air-equilibrated Buffer F constituted oxidizing conditions in this study. No further modifications to this buffering system were required to initiate the formation of any potential intersubunit disulfide bonds. The addition of freshly prepared 2–10 mM DTT followed by a minimum incubation of 6 h was sufficient to reduce all sulfhydryl crosslinks.
Urea denaturation profiles
PFK concentrations in all samples were adjusted to 20 μg/mL and incubated for at least 24 h in Buffer F containing the desired concentration of urea, and, when appropriate, 10 mM DTT. The percentage activity and fluorescence plotted in Figure 4 ▶ are relative to protein similarly incubated in the absence of urea. No more than a 2% loss in activity was noted in any of these controls.
Steady-state fluorescence measurements at atmospheric pressure
For urea dissociation profiles, intrinsic PFK fluorescence was monitored at 25°C with an SLM 4800 spectrofluorometer equipped with PX01 photon counting electronics and controlled via software obtained from ISS, Inc. The samples were excited at 300 nm, and emission was collected through a 2 mm-thick Schott WG-335 cut-on filter. All measurements were made in a 0.4-cm cuvette to minimize inner filter effects deriving from DTT absorbance, and were corrected for any background contribution to emission.
Fluorescence emission spectra for hybrids between E195,199K/W311Y and wild-type PFK were generated on the instrument described above, using 295 nm excitation. A vertically oriented polarizer was placed in the emission path to avoid grating-produced spectral aacts. All spectra were corrected for buffer signal contribution and normalized to constant protein concentration.
Steady-state fluorescence measurements under high hydrostatic pressure
All measurements were performed at 25°C using PFK concentrations of 25 μg/mL in Buffer F containing, when specified, 2 mM DTT. Fluorescence deriving from samples subjected to hydrostatic pressure in an ISS high-pressure spectroscopy cell was collected on an ISS Koala spectrofluorometer. The ∼0.8-mL sample was contained within a bottle cuvette sealed by flexible plastic tubing that served as a diaphragm to separate the sample from the pressurizing medium (ethanol) while transmitting the pressure to the sample. Pressure was generated and maintained with a computer-controlled, automated, screw-drive pump with a feedback pressure sensor obtained from Advanced Pressure Products. Samples were allowed to incubate for 15 min after each 100 bar pressure change prior to data collection. Fluorescence was monitored through a 2 mm-thick Schott WG-345 cut-on filter with excitation at 300 nm. Measurements of catalytic activity on samples subjected to 2400 bar were performed at least 12 h after depressurization.
Hybrid tetramer formation and their unambiguous ideication
Hybrid tetramers containing both wild-type and charge-tagged subunits were formed by mixing wild-type PFK and E195,199K in the presence of 0.4 M KSCN for 2 h followed by dialysis versus buffer F over 4 h to decrease the KSCN concentration to less than 1 mM. The various hybrids were separated with a Pharmacia (FPLC) Mono-Q anion exchange column using a linear NaCl gradient. Six to seven total peaks resulted, corresponding to all possible subunit combinations (4:0, 3:1, 2:2, 1:3, and 0:4), including up to three possible 2:2 hybrids (overlapping in their elution between 0.20 and 0.21 M NaCl), which differ only in the position of charge tag relative to the axes of symmetry (Fig. 6 ▶). Notably, the percentage of the total area associated with each peak (treating the 2:2 hybrids collectively) corresponds to the value predicted by combinatorial degeneracy, suggesting that subunits of charge-tagged mutants and wild-type PFK have very similar affinities for each other.
Fig. 6.

Differential elution of hybrid PFK tetramers from anion exchange. A mixture of hybrid tetramers between wild-type PFK and E195,199K were loaded onto a Mono-Q anion-exchange column and eluted with a linear NaCl gradient. The absorbance at 280 nm (solid line) of the eluent was continually monitored, and the enzymatic activity (dashed line) was assayed for each 0.5-mL fraction. Hybrid compositions were determined as described in the text and indicated within the figure in the form "E195,199K:wild-type PFK."
The identities of the peaks appearing in Figure 6 ▶ were confirmed in two ways. First, a W311Y mutation was engineered into the E195,199K mutant to remove the only tryptophan. Each hybrid tetramer between wild-type PFK and E195,199K/W311Y, separated via the Pharmacia Mono-Q anion-exchange column described above, was then individually assessed for subunit composition by its relative tryptophan fluorescence (Fig. 7 ▶). Upon normalization to constant protein concentration, the fluorescence deriving from samples of peaks 2, 3–5, and 6 approximately correspond to 25, 50, and 75%, respectively, of the emission from wild type (sample 7). Peaks 3–5 exhibited nearly identical emission spectra, suggesting that each peak represents a 2:2 combination of subunits. The first and last peaks of the profile elute at a concentration of NaCl corresponding to the elution profiles of E195,199K and wild-type PFK, respectively.
Fig. 7.

Normalized tryptophan emission spectra of hybrid tetramers between E195,199K/W311Y and wild-type PFK. Hybrids of E. coli PFK between wild-type subunits, which contain a single tryptophan, and charge-tagged subunits (E195,199K) containing the tryptophan deletion mutation (W311Y) were made and isolated as described in the text and in Figure 2 ▶. Fluorescence emission spectra (λex = 295 nm) were normalized to constant protein concentration. Ratios of subunits within each hybrid are indicated following the format "E195,199K/W311Y:wild type."
The second ideication procedure employed native PAGE (Laemmli 1970). The hybrid mixture produces in five bands (e.g., see Fig. 2 ▶, lanes 4,5). Peaks 1 and 7 from Figure 6 ▶ share identical mobilities with those of pure E195,199K and wild-type PFK, respectively. Likewise, peaks 3–5 comigrate on native gel electrophoresis equidistant from pure wild-type and pure mutant tetramers. Peaks 2 and 6 migrate at distances consistent with 3:1 and 1:3 hybrid combinations of charge-tagged and wild-type PFK.
The preparation of hybrids between E195,199K/W311Y and wild-type PFK differed somewhat from the methods outlined above because of the decreased stability of the tryptophan deletion mutant. These hybrids were generated by incubating separately 4 mg of each in Buffer F containing 0.4 M KSCN. Wild-type protein was exposed to KSCN for 60 min, whereas the mutant E195,199K/W311Y was exposed to the KSCN for only 30 min followed by mixing for 20 min in the presence of 1 mM Fru-6-P. The mixture was dialyzed at 4°C into Buffer F supplemented with 5% glycerol and 1 mM Fru-6-P. Samples were removed from glycerol and Fru-6-P via a desalting column prior to subsequent experiments.
SDS-PAGE analyses
Denaturing Laemmli (1970) electrophoresis utilizing 4% stacking/10% resolving gels were run at 200 V at room temperature for approximately 45 min. Samples treated with NEM were incubated at concentrations of 0.1 mg/mL in Buffer F containing 1 mM NEM at room temperature for at least 12 h prior to gel loading.
Linkage analyses
The notation employed to describe dissociation and coupling constants on E. coli PFK has been previously defined (Johnson and Reinhart 1992Johnson and Reinhart 1994a, 1997). In summary, the ligands Fru-6-P, MgATP, MgADP, and PEP are designated by the subscripts a, b, x, and y, respectively. In a modification of the notation of Cleland (1963), dissociation constants for Fru-6-P, MgADP, and PEP in the absence of other ligands are designated as K0ia, K0ix, and K0iy, respectively. If a dissociation constant reflects the presence of other bound ligands, the bound ligands will be denoted in the subscript following "/". For example, Kiy/b refers to the dissociation constant of PEP from PFK saturated with the substrate MgATP. The total influence one ligand exerts on the binding affinity of the second is quaied by the coupling constant, Q, defined below for the thermodynamic linkage between Fru-6-P and PEP in the saturating presence of the second substrate, MgATP:
 |
(2) |
Coupling parameters are experimentally determined by measuring the dependence of the apparent dissociation constant for one ligand on the concentration of the second ligand and fitting these data to equation 2 as described previously (Reinhart 1983Reinhart 1985Reinhart 1988; Johnson and Reinhart 1992 Johnson and Reinhart 1994a):
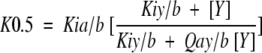 |
(3) |
The notation in equation 3 specifically pertains to the coupling between Fru-6-P and PEP when MgATP is maintained at a saturating concentration, but is easily modified to incorporate allosteric influences by MgADP, substituting X for Y in the notation. The dissociation constant for Fru-6-P is designated as K0.5 in equation 3, because Fru-6-P binding curves exhibit positive cooperativity under most conditions.
Acknowledgments
We thank Dr. Robert Kemp at the Chicago Medical School for originally providing our lab with the pRZ3 plasmid containing the E. coli PFK gene. We also thank Dr. Fabiola Janiak of the University of Oklahoma for her efforts in transferring the PFK gene into the pAlter plasmid for expression and subsequent genetic manipulation. Finally, we acknowledge the efforts of Anthony Burch, Southwestern Oklahoma State University, for his help in screening potential cross-linking mutations. This work was supported by the National Institutes of Health (GM33216 to GDR) and by the Robert A. Welch Foundation (A1368 to GDR).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
PFK, phosphofructokinase
SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel electrophoresis
NEM, N-ethyl maleimide
Fru-6-P, fructose 6-phosphate
Fru-1,6-BP, fructose 1,6-bisphosphate
PEP, phospho(enol)pyruvate
MOPS, 3-(N-morpholino)propanesulfonic acid
EDTA, ethylenediaminetetraacetic acid
LB, Luria-Bertaini
TRIS, 2-amino-2-(hydroxymethyl)-1,3-propanediol
BCA, bicinchoninic acid
EPPS, N-(2-hydroxyethyl)piperazine-N`-(3-propanesulfonic acid)
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.02401.
References
- Betz, S.F. 1993. Disulfide bonds and the stability of globular proteins. Protein Science 2 1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangy, D., Buc, H., and Monod, J. 1968. Kinetics of the allosteric interactions of phosphofructokinase from Escherichia coli. J. Mol. Biol. 31 13–35. [DOI] [PubMed] [Google Scholar]
- Cleland, W.W. 1963. The kinetics of enzyme-catalyzed reactions with two or more substrates of products. I. Nomenclature and rate equations. Biochim. Biophys. Acta 67 104–137. [DOI] [PubMed] [Google Scholar]
- Daldal, F. 1983. Molecular cloning of the gene for phosphofructokinase-2 of Escherichia coli and the nature of a mutation, pfkB1, causing a high level of the enzyme. J. Mol. Biol. 168 285–305. [DOI] [PubMed] [Google Scholar]
- Deville-Bonne, E. and Else, A.J. 1991. Reversible high hydrostatic pressure inactivation of phosphofructokinase from Escherichia coli. Eur. J. Biochem. 200 747–750. [DOI] [PubMed] [Google Scholar]
- Deville-Bonne, D., Le Bras, G., Teschner, W., and Garel, J.-R. 1989. Ordered disruption of subunit interfaces during the stepwise reversible dissociation of Eschericia coli phosphofructokinase with KSCN. Biochemistry 28 1917–1922. [DOI] [PubMed] [Google Scholar]
- Flory, P.J. 1956. Theory of elastic mechanisms in fibrous proteins. J. Am. Chem. Soc. 78 5222–5235. [Google Scholar]
- Hellinga, H.W. and Evans, P.R. 1985. Nucleotide sequence and high-level expression of the major Escherichia coli phosphofructokinase. Eur. J. Biochem. 149 363–373. [DOI] [PubMed] [Google Scholar]
- Heremans, K. and Smeller, L. 1998. Protein structure and dynamics at high pressure. Biochim. Biophys. Acta 1386 353–370. [DOI] [PubMed] [Google Scholar]
- Johnson, J.L. and Reinhart, G.D. 1992. MgATP and fructose 6-phosphate interactions with phosphofructokinase from Escherichia coli. Biochemistry 31 11510–11518. [DOI] [PubMed] [Google Scholar]
- ———. 1994a. Influence of MgADP on phosphofructokinase from Escherichia coli elucidation of coupling interactions with both substrates. Biochemistry 33 2635–2643. [DOI] [PubMed] [Google Scholar]
- ———. 1996. High pressure effects on E. coli phosphofructokinase. In High pressure effects in molecular biophysics and enzymology (eds. J.L. Markley, C.A. Royer, and D.B. Northrop), Oxford University Press, New York, pp. 242–255.
- ———. 1997. Failure of a two-state model to describe the influence of phospho(enol)pyruvate on phosphofructokinase from Escherichia coli. Biochemistry 36 12814–12822. [DOI] [PubMed] [Google Scholar]
- Kotlarz, D. and Buc, H. 1977. Two Escherichia coli fructose-6-phosphate kinases: Preparative purification, oligomeric structure, and immunological studies. Biochem. Biophys. Acta 484 35–48. [DOI] [PubMed] [Google Scholar]
- ———. 1982. Phosphofructokinases from Escherichia coli. Methods Enzymol. 90 60–70. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Le Bras, G., Auzat, I., and Garel, J.-R. 1995. Tetramer-dimer equilibrium of phsphofructokinase and formation of hybrid tetramers. Biochemistry 34 13203–13210. [DOI] [PubMed] [Google Scholar]
- Le Bras, G., Teschner, W., Deville-Bonne, D., and Garel, J.-R. 1989. Urea-induced inactivation, dissociation, and unfolding of the allosteric phosphofructokinase from Escherichia coli. Biochemistry 28 6836–6841. [DOI] [PubMed] [Google Scholar]
- Reinhart, G.D. 1983. The determination of thermodynamic allosteric parameters of an enzyme undergoing steady-state turnover. Arch. Biochem. Biophys. 225 389–401. [DOI] [PubMed] [Google Scholar]
- ———. 1985. Influence of pH on the regulatory kinetics of rat liver phosphofructokinase: A thermodynamic linked-function analysis. Biochemistry 24 7166–7172. [DOI] [PubMed] [Google Scholar]
- ———. 1988. Linked-function origins of cooperativity in a symmetrical dimer. Biophys. Chem. 30 159–172. [DOI] [PubMed] [Google Scholar]
- Schirmer, T. and Evans, P.R. 1990. Structural basis of the allosteric behavior of phosphofructokinase. Nature 343 140–145. [DOI] [PubMed] [Google Scholar]
- Shirakihara, Y. and Evans, P.R. 1988. Crystal structure of the complex of phosphofructokinase from Escherichia coli with its reaction products. J. Mol. Biol. 204 973–994. [DOI] [PubMed] [Google Scholar]
- Teschner, W. and Garel, J.-R. 1989. Intermediates on the reassociation pathway of phosphofructokinase I from Escherichia coli. Biochemistry 28 1912–1916. [DOI] [PubMed] [Google Scholar]
- Weber, G. 1972. Ligand binding and internal equilibria in proteins. Biochemistry 11 864–878. [DOI] [PubMed] [Google Scholar]
- ———. 1975. Energetics of ligand binding to proteins. Adv. Protein Chem. 29 1–83. [DOI] [PubMed] [Google Scholar]
- Xu, J., Oshima, T., and Yoshida, M. 1990. Tetramer-dimer conversion of phosphofructokinase from Thermus thermophilus induced by its allosteric effectors. J. Mol. Biol. 215 597–606. [DOI] [PubMed] [Google Scholar]
- Zheng, R.-L. and Kemp, R.G. 1992. The mechansim of ATP inhibition of wild type and mutant phosphofructo-1-kinase from Escherichia coli. J. Biol. Chem. 267 23640–23645. [PubMed] [Google Scholar]