

Abstract
Streptomyces venezuelae synthesizes chloramphenicol (Cm), an inhibitor of ribosomal peptidyl transferase activity, thereby inhibiting bacterial growth. The producer escapes autoinhibition by its own secondary metabolite through phosphorylation of Cm by chloramphenicol phosphotransferase (CPT). In addition to active site binding, CPT binds its product 3-phosphoryl-Cm, in an alternate product binding site. To address the mechanisms of Cm tolerance of the producer, the crystal structures of CPT were determined in complex with either the nonchlorinated Cm (2-N-Ac-Cm) at 3.1 Å resolution or the antibiotic's immediate precursor, the p-amino analog p-NH2-Cm, at 2.9 Å resolution. Surprisingly, p-NH2-Cm binds CPT in a novel fashion. Additionally, neither 2-N-Ac-Cm nor p-NH2-Cm binds to the secondary product binding site.
Keywords: Antibiotic, chloramphenicol, kinase, phosphorylation, resistance
Streptomycetes are gram-positive soil bacteria that have complex life cycles and produce a range of secondary metabolites, most of which are biologically active and often medicinally useful. Indeed, more than 60% of the known antibiotics are produced by the species of Streptomyces. Streptomyces venezuelae ISP5230 produces chloramphenicol (Cm; Fig. 1 ▶) from chorismic acid in a complex pathway that includes eight biochemically interesting steps (Vining and Westlake 1984). The detailed sequence of events after formation of p-aminophenylserine remains unclear; however, oxidation of acrylamine to form Cm is the final step in the pathway (Fig. 1 ▶). Although the mode of incorporation of the two chlorine atoms is unknown, the isolation of the N-acetyl Cm analog from S. venezuelae cml− strains suggests that an N-acyl precursor of Cm is the substrate for chlorination (Doull et al. 1985).
Fig. 1.

Structure of Cm and Cm-analogs. CPT uses ATP as the phosphoryl donor to transfer the γ-phosphate to the primary (C-3) hydroxyl of Cm (resulting in 3-phosphoryl-Cm). An amino group replaces the nitro group in the immediate precursor of the Cm pathway, α-N-dichloroacetyl-p-aminophenylserinol (p-NH2-Cm). In the nonchlorinated Cm (boxed) N-acetyl-p-nitro-phenylserinol (2-N-Ac-Cm), hydrogens replace the two chlorines.
The bacteriostatic activity of Cm results from its binding to 50S prokaryotic ribosomes at a site that blocks the peptidyl transferase step in protein biosynthesis (Vining and Westlake 1984). Virtually all of the chemical functions of Cm contribute to its effectiveness as an inhibitor. Therefore, it is not surprising that enzyme-mediated resistance to Cm has been reported to arise from dehalogenation, nitro group reduction, hydrolysis of the amide bond, and modification of one or both of the hydroxyls of Cm by either phosphorylation or acetylation of the antibiotic (Pongs 1979; Waring et al. 1981; Shaw 1983; 1992; Tennigkeit and Matzura 1991). Chloramphenicol acetyltransferase (CAT), which is found in some bacteria but not in the producer, attaches an acetyl group to the C-3` hydroxyl, thereby forming an inactive product (Shaw and Leslie 1991). Because of its toxic effects on hematopoiesis, Cm is used only to treat life-threatening infections. Analogs of Cm that lack the aromatic NO2 group, which is thought to cause irreversible marrow aplasia, have been developed.
S. venezuelae evades the toxicity of its own lethal metabolite because chloramphenicol phosphotransferase (CPT) phosphorylates the primary (C-3) hydroxyl of Cm (Mosher et al. 1995) to provide tolerance (resistance). CPT may also phosphorylate the immediate precursor of Cm, the p-amino analog α-N-dichloroacetyl-p-aminophenylserinol (p-NH2-Cm; Fig. 1 ▶), which would put the resistance (tolerance) mechanism in place before oxidation of the p-amino substituent to a nitro group generates an autoinhibitory agent. However, the CTP–p-NH2-Cm crystal structure described here suggests that p-NH2-Cm binds to the CPT active site but is not a substrate for the enzyme.
The recently determined crystal structure of CPT in complex with bound substrates revealed its catalytic mechanism: Asp 37 accepts a proton from the 3-hydroxyl group of Cm concurrently with nucleophilic attack of the resulting oxyanion on the γ-phosphate of ATP (Izard and Ellis 2000). A distinct alternate product binding site was found 13 Å away from the active site. To address the tolerance mechanism in S. venezuelae, the three-dimensional structures of CPT were determined in complex with p-NH2-Cm and in complex with the nonchlorinated Cm (2-N-Ac-Cm).
Results
The binding of nonclorinated Cm (2- N-Ac-Cm) to CPT
The strongest feature of the difference maps calculated from crystals of CPT in complex with 2-N-Ac-Cm is found within the Cm-binding pocket (Fig. 2A ▶). Strong electron density is clearly visible for the entire molecule of the ligand; however, no electron density is present for the corresponding chlorine atoms of Cm. Indeed, the deepest hole in the Fo − Fc electron density map with a negative peak height of >6–8 times the noise level was found for the two chlorine atoms when Cm was included in the model instead of 2-N-Ac-Cm (not shown). The binding of 2-N-Ac-Cm to the enzyme is shown in Figures 3A and 4A ▶ ▶. Unlike the CPT structures in complex with Cm, no electron density was found in the alternate product binding site (i.e., only one 2-N-Ac-Cm molecule was bound per protomer). The lack of chlorine in 2-N-Ac-Cm does not create a cavity as the chlorines in Cm point toward the solvent channels within the crystal lattice.
Fig. 2.

Stereodiagram of the CPT protomer structure showing electron density for (A) 2-N-Ac-Cm and (B) p-NH2-Cm. The final σA weighted Fo − Fc omit electron density maps are contoured at 3.5σ at (A) 3.1 Å resolution and (B) 2.9 Å resolution. The antibiotic bound to the alternate product binding site and the nucleotide found in the CPT–ATPγS–Cm ternary complex are superimposed onto the CPT–2-N-Ac-Cm structure in A. This figure, along with Figure 4 ▶, was produced with BobScript (Kraulis 1991) and Raster3D (Merritt and Bacon 1997).
Fig. 3.
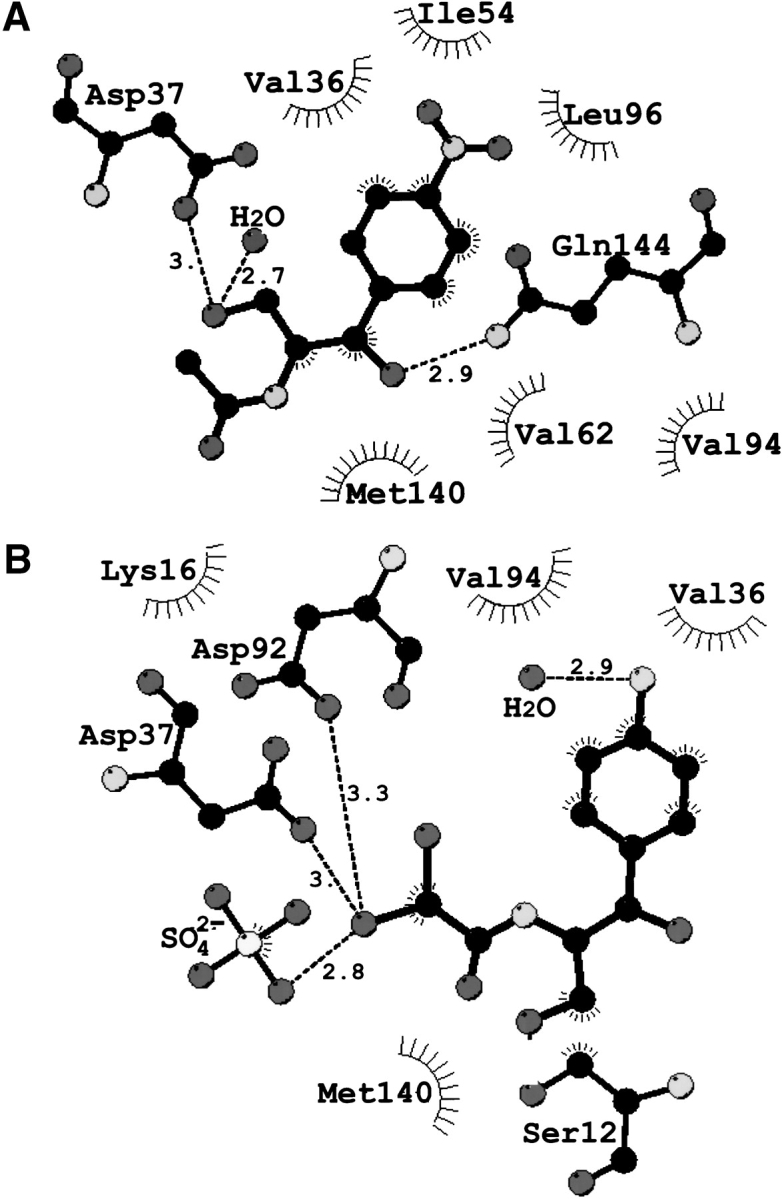
Schematic diagram of the principal interactions made by (A) 2-N-Ac-Cm and (B) p-NH2-Cm in the binary complex with CPT. (Broken lines) Hydrogen-bonding interactions. Distances are indicated in angstrom units between two electronegative atoms. (Black) Carbon atoms; (dark gray) oxygen and chlorine atoms; and (light gray) nitrogen atoms. This figure was produced with LIGPLOT (Wallace et al. 1995).
Fig. 4.

Close-up view of the density in the ligand-binding sites. The final σA weighted Fo − Fc omit electron density maps are contoured at 3.5σ at 3.1 Å resolution (A) and 2.9 Å resolution (B). (A) CPT–2-N-Ac-Cm (black) is superimposed onto CPT–Cm (gray), and (B) CPT–p-NH2-Cm (black) is superimposed onto CPT–ATPγS–Cm (gray). The 2-N-Ac-Cm buries 418 Å2 of solvent-accessible surface area on binding and p-NH2-Cm occludes 451 Å2 on binding. The label of the catalytic base (Asp 37) abstracting a proton from the hydroxyl of Cm is boxed. The oxygen of the reactive hydroxyl is drawn as a sphere. (B) The water molecule found in the CPT– p-NH2-Cm structure (interacting with the p-amino group of the ligand) refined to a temperature factor of 50 Å2 and was additionally omitted in the shown omit map. The viewing orientation of this figure differs from that of Figure 3 ▶.
The binding of p-NH2-Cm to CPT
The crystal structure of CPT in complex with the immediate precursor of Cm shows that p-NH2-Cm binds within the Cm-binding pocket (Fig. 2B ▶). A water molecule forms hydrogen bonds with the amide group (Figs. 3B and 4B ▶ ▶) and resides 3.8 Å (4 Å) from the carboxylate group of Asp 93 (Glu 72). The ligand's primary hydroxyl group is oriented away from the proposed catalytic Asp 37 and instead engages in a hydrogen bonding interaction to the side chain of Ser 12. The chlorine atoms stretch into the ATP-binding pocket and occlude the γ-phosphate binding site of ATP. The halogenated group interacts with a sulfate anion found at the β-phosphate-binding site of ATP. Novel hydrophobic interactions between the p-amino analog of Cm and CPT inovlve Lys 16 and Asp 92.
The poorest parts of the refined CPT–p-NH2-Cm model are two regions comprising flexible loops where model building was difficult because of weak density: Temperature factors of residues 49–51 located on the loop immediately following α-helix 3 (α3; Fig. 2 ▶) and residues 57–60 located on the loop before α4 refined only to values that were comparable to the protein structure if an occupancy of one half was used. As was the case for 2-N-Ac-Cm bound to the enzyme, no electron density was found in the alternate product binding site in the CPT–p-NH2-Cm structure.
Discussion
The observation that p-NH2-Cm binds the enzyme in a distinct orientation within the active-site pocket was unexpected. In particular, because both the nitro group of Cm and the amino group of p-NH2-Cm are exposed to the solvent. Therefore, these groups do not engage in extensive interactions with the enzyme. Distinctly, one water molecule is within hydrogen-bonding distance (2.9 Å) to the amide group of p-NH2-Cm but is not present in the CPT–Cm structure. Moreover, only very small conformational changes are required by the protein to bind either Cm or p-NH2-Cm in its novel fashion. For example, the side chains of Val 62 and Leu 96 are further away from the ligand in the CPT–p-NH2-Cm structure (0.65 Å and 0.43 Å, respectively), whereas Val 94 is now closer to the modified antibiotic (0.6 Å). The benzyl ring of p-NH2-Cm bound to CPT is rotated by about 45° with respect to the benzyl rings of Cm or 2-N-Ac-Cm seen in the CPT–Cm, CPT–ATPγ–Cm, and CPT–2-N-Ac-Cm structures. The remainder of p-NH2-Cm is rotated almost 120° away from Cm's binding position, thereby pointing its chlorine atoms well into the ATP-binding pocket. This arrangement places the primary alcohol group of p-NH2-Cm 6 Å away from the proposed catalytic Asp 37. Thus, the structure of CPT–p-NH2-Cm suggests that the immediate precursor of Cm cannot be phosphorylated by CPT. Interestingly, the p-amino precursor also cannot be acetylated by CAT.
The overall protein structure of CPT in complex with Cm analogs is very similar to that of the enzyme in the absence or presence of nucleotide, or both nucleotide and Cm (Izard and Ellis 2000). The overall root mean squares deviation (rmsd) for the Cα positions of all residues (1–178) of the CPT–p-NH2-Cm (or CPT–2-N-Ac-Cm) structure superimposed onto the substrate-free CPT crystal structure is 0.3 Å (0.46 Å). The 178 Cα atoms of CPT–p-NH2-Cm superimpose onto the CPT–2-N-Ac-Cm structure with an overall rmsd of 0.5 Å. Discrepancies of >3.5 Å in Cα positions occur for residues 135–137, which are located on loop α6–α7 of the lid domain. Arg 136 moves during catalysis and promotes bond cleavage by stabilizing the developing negative charge on the equatorial oxygen atom of the γ-phosphate in the transition state. The conformation of Arg 136 in the CPT–p-NH2-Cm structure resembles that in the substrate-free CPT structure and significantly differs from that in the enzyme bound to Cm, nucleotide, or both Cm and nucleotide. Previous crystallographic analyses determined that Arg 136 is almost within hydrogen-bonding distance of the reactive hydroxyl of Cm and the γ-phosphate of the nucleotide. The novel binding mode of p-NH2-Cm places the reactive hydroxyl in a position where it cannot form a hydrogen bond with Arg 136. The findings of the present study are consistent with a movement of Arg 136 occurring once either substrate (Cm or ATP) binds and support the previously suggested domain closure on substrate binding as also seen in other kinases.
Superposition of the previously determined structures of substrate-free enzyme, CPT–ATP, CPT–Cm, and CPT–ATPγS–Cm onto the CPT–p-NH2-Cm or CPT–2-N-Ac-Cm structures described here also highlights a movement of up to 2 Å for the backbone of residues 50–52 located at the alternate product binding site. In particular, the side chain of Glu 51 displays a variety of conformations amongst the six structures. The largest shift occurs for superposition of residues 50–52 of CPT–p-NH2-Cm onto any of the other CPT structures. However, it is difficult to determine which interaction(s) and residue(s) are critical for the binding of the product resembling Cm–SO42− (as bound to the enzyme in the CPT–Cm and CPT–ATPγS–Cm crystal structures) but not for the binding of p-NH2-Cm and 2-N-Ac-Cm at the alternate product binding site. The lack of binding Cm or its derivatives at the alternate product binding site supports the suggested biological role of the alternate product binding site, whereby CPT acts as a carrier and facilitates the export of inactivated, phosphorylated antibiotic (but not the p-amino analog or nonhalogenated forms of Cm) to an efflux pump. Therefore, the efflux protein may export Cm-3`-phosphate, but not the antibiotic's derivatives. The protective phosphate group is easily removed by an extracellular phosphatase, thus releasing of Cm to foreign organisms in the producer's vicinity.
These findings raise new questions regarding the co-evolution of the biosynthetic pathway of the antibiotic and the protective mechanisms of its producer. In addtion, strong evidence indicates that there are multiple mechanisms for resistance to Cm, just as there are multiple mechanisms for resistance to tetracycline, daunorubicin, erythromycin, and various other antibiotics in the producing organisms.
Materials and methods
Crystallization and data collection
Crystallization experiments were performed at room temperature using the hanging-drop vapor-diffusion technique. The purified enzyme was dialyzed into 50 mM Tris/HCl (pH 7.5) containing 0.4 M ammonium sulphate, and concentrated to 7.5 mg/mL. CPT crystals with bound ligands were obtained from 0.1 M Tris/HCl (pH 7), 1.6 M ammonium sulphate, 4% acetonitrile, and 2 mM ligand. Crystallization droplets of 10 μL resulted in CPT cocrystals of dimensions up to (0.4 mm)3 after about 2 wk. Crystals were cryoprotected by inclusion of 30% glycerol into the mother liquor and were shown to belong to space group I4132 (a = 200.0 Å), with one polypeptide chain in the asymmetric unit, a solvent content of 0.86 and a volume-to-protein-mass ratio (Vm) of 8.8 Å3 dalton (Ellis et al. 1999). X-ray data of CPT–p-NH2-Cm were collected at 100 K to 2.9 Å resolution on a CCD detector at beam lin 9.6 at SRS, Daresbury, with the wavelength set to 0.87 Å. X-ray data of CPT–2-N-Ac-Cm were collected at 100 K to 3.1 Å resolution on a CuKα rotating anode on an RAXIS-4 imaging plate. All data were processed with DENZO and SCALEPACK (Otwinowski & Minor 1997). Data statistics are given in Table 1.
Table 1.
Crystallographicdataandrefinementstatistic
| Data collection | CPT:2-N-Ac-Cm | CPT:p-NH2-Cm |
| Resolution [Å] | 3.1 | 2.9 |
| Total data | 229,617 | 370,300 |
| Unique data | 12,472 | 14,719 |
| Overall completeness | 0.98 | 0.951 |
| Completeness (last shell) | 0.906 | 0.593 |
| F2 > 3σ(F2) [%] | 84.6 | 77.8 |
| Ramerge (overall) | 0.167 | 0.051 |
| Ramerge (last shell) | 0.323 | 0.298 |
| Average F2/σ(F2) | 13.9 | 48.8 |
| Crystallographic refinement | ||
| Number of reflections | 10,677 | 14,463 |
| Final model parameters | ||
| Number of amino acid residues | 178 | 178 |
| Number of protein atoms | 1320 | 1320 |
| Number of solvent molecules | 81 | 92 |
| Resolution range [Å] | 42 - 3.1 | 45 - 2.9 |
| R-factorb (overall) | 0.226 | 0.278 |
| R-factorb (last shell) | 0.336 | 0.438 |
| Rcfree (overall) | 0.256 | 0.297 |
| Rcfree (last shell) | 0.332 | 0.463 |
| Average main-chain B-factor [Å2] | 40.2 | 60.4 |
| Average side-chain B-factor [Å2] | 51.4 | 67.7 |
| Average water molecule B-factor [Å2] | 35.29 | 56.4 |
| Average ligand B-factor [Å2] | 72.8 | 79.7 |
| r.m.s. deviations from ideal geometry | ||
| Covalent bond lengths [Å] | 0.006 | 0.008 |
| Bond angles [˚] | 1.1 | 1.3 |
 |
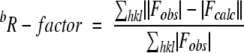 |
c The free R-factor is a cross-validation residual calculated by using 5% of the native data which were randomly chosen and excluded from the refinement.
Structure determination and crystallographic refinement
The final model (including water molecules) refined against data of the inflection point of the fluorescence of the seleno-methionyl CPT isoform (Izard and Ellis 2000) was used as a starting model for refinement. Both CPT structures were refined by use of standard protocols for the program CNS (Brünger et al. 1987; 1998). The free R-value (Brünger 1992) was monitored throughout the refinement. Additional water molecules were initially identified in Fo − Fc maps and screened for reasonable geometry and for a refined thermal factor <50 Å2 in the CPT–2-N-Ac-Cm structure and <80 Å2 in the CPT–p-NH2-Cm structure. Table 1 shows the overall crystallographic R-factor and the free R-factor for all models for all observed reflections within the indicated resolution range. A Ramachandran plot analysis of both structures by the program PROCHECK (CCP4 1994) indicated that 90.7%–91.3% of all the residues lie in most favorable regions and that 8.7%–9.3% lie in additional allowed regions. This structure analysis also showed that all stereochemical parameters were better than expected at the given resolution.
Acknowledgments
The author thanks Angela McArthur (SJCRH) for editing the manuscript, Jackie Ellis (Leicester) for expert assistance with biochemistry and crystallization, and Bill Shaw (Leicester) for initiating the project. This work was supported in part by Cancer Center Support (CORE) Grant CA 21765 and by the American Lebanese Syrian Associated Charities (ALSAC).
This work was supported by National Institutes of Health Grant no. EY08918, and in part by Fight for Sight, Inc., and Research to Prevent Blindness, Inc.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.430101.
References
- Brünger, A.T. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Kuriyan, J., and Karplus, M. 1987. Crystallographic R- factor refinement by molecular dynamics. Science 235 458–460. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kunszeweski, J., Nilges, M., Pannu, M.S., et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallograph D54 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallograph. D50 760–763. [DOI] [PubMed] [Google Scholar]
- Doull, J., Ahmed, Z., Stuttard, C., and Vining, L.C. 1985. Isolation and characterization of Streptomyces venezuelae mutants blocked in chloramphenicol biosynthesis. J. Gen. Microbiol. 131 97–104. [DOI] [PubMed] [Google Scholar]
- Ellis, J., Campopiano, D.J., and Izard, T. 1999. Cubic crystals of chloramphenicol phosphotransferase from Streptomyces venezuelae in complex with chloramphenicol. Acta Crystallograph. D55 1086–1088. [DOI] [PubMed] [Google Scholar]
- Izard, T. and Ellis, J. 2000. The crystal structures of chloramphenicol phosphotransferase reveal a novel inactivation mechanism. EMBO J. 19 2690–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallograph. 24 946–950. [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Mosher, R.H., Camp, D.J., Yang, K., Brown, M.P., Shaw, W.V., and Vining, L.C. 1995. Inactivation of chloramphenicol by O-phosphorylation. J. Biol. Chem. 270 27000–27006. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pongs, O. 1979. Chloramphenicol. In Mechanism of action of antibacterial agents pp. 26–42. Springer, Berlin.
- Shaw, W.V. 1983. Chloramphenicol acetyltransferase: Enzymology and molecular biology. Crit. Rev. Biochem. 14 1–46. [DOI] [PubMed] [Google Scholar]
- ———. 1992. Chemical anatomy of antibiotic resistance: Chloramphenicol acetyltransferase. Sci. Prog. 76 565–580. [PubMed] [Google Scholar]
- Shaw, W.V. and Leslie, A.G.W. 1991. Chloramphenicol acetyltransferase. Annu. Rev. Biophys. Chem. 20 363–386. [DOI] [PubMed] [Google Scholar]
- Tennigkeit, J. and Matzura, H. 1991. Nucleotide sequence analysis of a chloramphenicol-resistance determinant from Agrobacterium tumefaciens and identification of its gene product. Gene 98 113–116. [DOI] [PubMed] [Google Scholar]
- Vining, L.C. and Westlake, D.W.S. 1984. In Bio/technology of Industrial Antibiotics (ed. E.J. Vandamore), pp. 387–409. Marcel Dekker, New York, NY.
- Wallace, A.C., Laskowski, R.A., and Thornton, J.M. 1995. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Prot. Eng. 8 127–134. [DOI] [PubMed] [Google Scholar]
- Waring, M.J., Richmond, M.H., Reynolds, P.E., Cundliffe, E., and Gale, E.F. 1981. The molecular basis of antibiotic action 2 pp. 462–468. Wiley & Sons Inc., London.