

Abstract
Transthyretin (TTR) subunits were labeled with a charge-modifying tag to evaluate the possibility of subunit exchange between tetramers under physiological conditions. Starting with a mixture of two TTR homotetramers, one having all subunits tagged at the N termini and the other composed of untagged subunits, heterotetramer formation as a function of time and temperature was evaluated using ion exchange chromatography. The data indicate that the subunit exchange can occur under native conditions at physiological pH in vitro, albeit slowly. Wild-type TTR exchanges subunits on a timescale of days at 37°C and on a timescale of hours at 4°C. The familial amyloid polyneuropathy-associated variant V30M exchanges subunits at the same rate as wild-type TTR at 4°C but slower and less efficiently at 37°C. Small molecule tetramer stabilizers abolish TTR subunit exchange, supporting a dissociative mechanism.
Keywords: Transthyretin, amyloid, familial amyloid polyneuropathy, ion exchange chromatography
Transthyretin (TTR) is a homotetrameric 55-kD protein found in plasma and cerebrospinal fluid that functions to transport thyroid hormones and the retinol/retinal-binding protein complex. Dissociation of tetrameric TTR under mildly acidic conditions forms a structurally rearranged monomeric intermediate (Lai et al. 1996; Liu et al. 2000) capable of self-assembling into amyloid fibrils (Colon and Kelly 1992) that are indistinguishable from the extracellular deposits found in amyloid patients. Wild-type TTR composes the fibrils in senile systemic amyloidosis patients (SSA), whereas deposition of one of >70 TTR variants (with increased propensity to form fibrils) is associated with familial amyloid polyneuropathy (FAP; Saraiva 1995).
The vast majority of patients suffering from one of the more severe familial forms of TTR-associated amyloid disease are heterozygous for the FAP mutation. Therefore, wild-type and variant subunits of TTR are expected to be co-expressed in the liver, and the resulting mixed tetramers are secreted into the plasma. Five different TTR tetramers are expected, composed of one or two different types of subunits in a distribution that may either be statistical (1:4:6:4:1) or, more likely, governed by thermodynamic stability. V30M is the most common mutation associated with FAP, a disease that is always progressive and fatal (Holmgren et al. 1988). There has been no effective treatment besides liver transplantation, which replaces the mutant TTR gene by wild type, resulting in amyloid regression (Holmgren at al. 1993).
In this study, we aimed to evaluate whether subunit exchange occurs between TTR tetramers after they are secreted into the plasma from the liver, that is, under physiological conditions. The formation of TTR heterotetramers has been suggested in previous studies in which the mixing of rhesus monkey TTR with human TTR revealed hybridization, even though the tetrameric nature of TTR had not yet been established (Bernstein et al. 1970). Hybrid tetramer formation between mouse TTR and human wild-type or V30M TTR has been observed only after denaturation and coreconstitution of both proteins (Kohno et al. 1997). In both studies, analysis of hybrid formation was accomplished by native gel electrophoresis.
Our strategy of separating TTR heterotetramers used a wild-type TTR variant that was N-terminally labeled with a tandem Flag tag (FT2), comprising the sequence DYKDDDDKDYKDDDDK. FT2 wild-type TTR was mixed with untagged homotetrameric wild type (or V30M TTR), and the subunit exchange was monitored by ion exchange chromatography as a function of incubation time at both 4°C and 37°C. Experiments were performed at physiological TTR concentration (3.6 μM) at pH 7.4. Control experiments were performed to show that the FT2 does not modify the thermodynamics, kinetics, or amyloidogenicity of TTR.
Results
Denaturation studies show nearly identical thermodynamic stabilities of homotetrameric FT2 wild-type TTR and wild-type TTR
Curves reflecting denaturation in GdmSCN show virtually superimposable unfolding profiles for FT2 wild-type TTR and wild-type TTR (Fig. 1A ▶). The data indicate that FT2 wild-type TTR and wild-type TTR undergo similar structural changes in chaotropic solution with midpoints of denaturation (D½) at 1.04 M and 1.06 M, respectively. This data, when considered in combination with the nearly identical m-values, indicate that the thermodynamic stability of TTR is unaffected by the FT2.
Fig. 1.
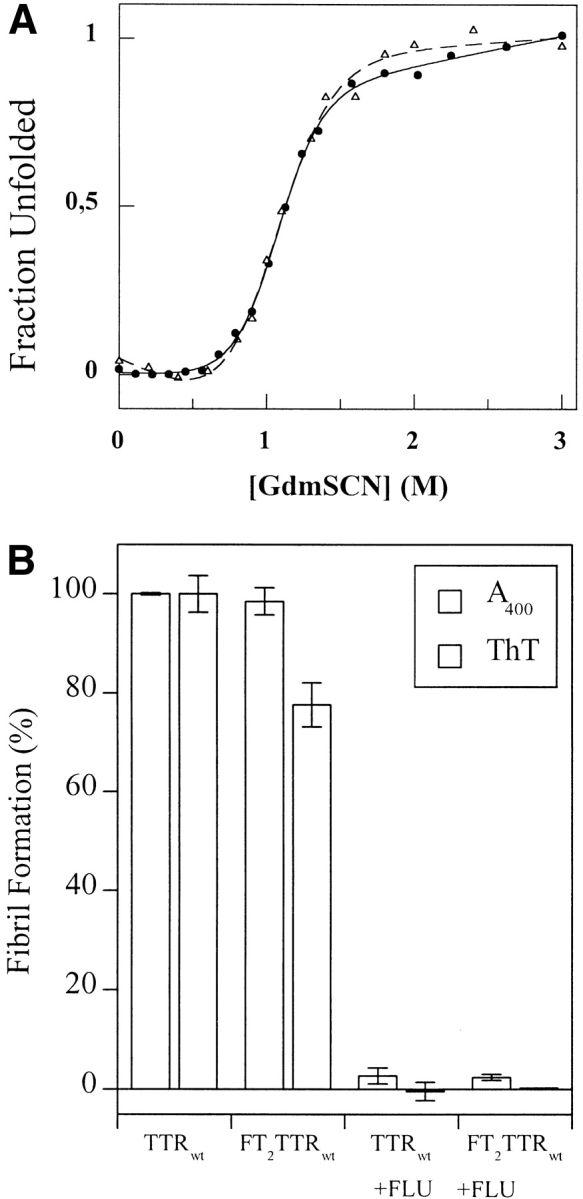

(A) Denaturation curves of FT2 wild-type TTR (open triangles, dashed curve) and wild-type TTR (filled circles, solid line) homotetramers recorded by the change in tryptophan fluorescence as a function of GdmSCN concentration. (B) Relative yield of amyloid fibrils produced from FT2 wild-type TTR and wild-type TTR (3.6 μM at pH 4.4) in presence and absence of inhibitor (7.2 μM flufenamic acid [FLU]) after incubation at 37°C for 72 h. (C) Sedimentation equilibrium analysis of fully exchanged FT2 wild-type TTR and wild-type TTR (7.2 μM at pH 7.4, exchanged for 5 d at 4°C). The line represents a fitted curve to a single ideal species model providing a MW of 55,722 ± 738 Da. The insert shows the distribution of random residuals.
Amyloidogenicity of FT2 wild-type TTR and wild-type TTR is very similar
A comparison of FT2 wild-type TTR and wild-type TTR reveals that their amyloidogenicities are very similar as discerned by turbidity and thioflavin T fluorescence analyses (Fig. 1B ▶). Fibril formation derived from either homotetrameric FT2 wild-type TTR or wild-type TTR is inhibited identically by the inhibitors flufenamic acid (Fig. 1B ▶) or thyroxine (data not shown). These small molecules occupy the two ligand-binding sites in TTR, resulting in tetramer stabilization (Petrassi et al. 2000), indicating that the binding sites within each tetramer are accessible and hence unaffected by the FT2. Amyloid formation from a heterogeneous mixture of tetramers prepared by incubation at 4°C for 1 wk is inhibited by the small molecules to the same extent (data not shown), showing that the thyroid hormone-binding sites are intact in the mixed tetramers prepared in this fashion.
Studies showing that the products of native subunit exchange are TTR tetramers
To further show that hybrid tetramers are formed upon incubation of FT2 wild-type TTR and wild-type TTR after 5 d at 4°C, sedimentation equilibrium studies were performed to rule out formation of other quaternary structures (Fig. 1C ▶). The data fit to a single ideal species model, showing random residuals with a solution molecular weight of 55,722 ± 738 Da, in very good agreement with the expected average molecular weight of TTR hybrid tetramers resulting from subunit exchange (between 55,600 Da [wild-type]4 and 63,600 [FT2-wild-type]4). Typically, the wild-type TTR homotetramer fits to an experimental molecular weight of 54400 Da (Lai et al. 1996).
Quantification of the composition of each hybrid tetramer formed by subunit exchange and preparatively separated by ion exchange chromatography was performed by RP-HPLC analysis using solvent conditions that dissociate the tetramers into subunits. The subunit stoichiometry correlated with the expected composition from retention on ion exchange chromatography (Fig. 2 ▶). The retention time on the anion ion exchange column (large chromatographic trace; Fig. 2 ▶) is proportional to the number of FT2 wild-type subunits: peak 1 is comprised of four untagged TTR subunits, and peak 5 is comprised of four FT2 wild-type subunits. Peaks 2, 3, and 4 have a 3:1; 2:2, and 1:3 wild-type:FT2 wild-type subunit stoichiometry, respectively. The retention time on the RP-HPLC column (small chromatograms) is reduced for the FT2 wild-type subunit because of the hydrophilic character of its polypeptide chain.
Fig. 2.

Chromatogram from ion exchange column at pH 8.0 (larger chromatogram with peaks 1–5) showing separation of hybrid TTR tetramers of FT2 wild-type TTR and wild-type TTR. The retention time increases proportionally with the number of FT2-subunits. After complete subunit exchange, five hybrid tetramers are afforded, containing no FT2-subunits (peak 1), one FT2-subunit (peak 2), two FT2-subunits (peak 3), three FT2-subunits (peak 4), and four FT2-subunits (peak 5). The eluted peaks from the ion exchange column were analyzed by RP-HPLC (small chromatograms) under conditions, which dissociated the tetramers into subunits to identify the TTR subunits composing the hybrid tetramers. The retention time in the RP-HPLC is shorter for the FT2-wild-type subunits because of the additional hydrophilic FT2 (peak A) in comparison with wild-type TTR (peak B).
The RP-HPLC peaks were identified as FT2 wild-type TTR (Fig. 2 ▶ (peak A)) or wild-type TTR (Fig. 2 ▶ (peak B)) by determining their masses using ESI-MS analysis. Peaks A and B showed masses of 15,876 and 13,890 Da, respectively.
TTR hybrid tetramers can form under native conditions
Using mixtures of FT2 wild-type TTR and wild-type TTR, it was possible to monitor subunit exchange between tetramers under nondenaturing conditions, using ion exchange chromatography. Figure 3A ▶ shows the time dependence of wild-type TTR subunit exchange initiated by mixing FT2 wild-type TTR and wild-type TTR homotetramers at 4°C. The temperature dependence of the rate of subunit exchange is illustrated in Figure 3B ▶, showing that the kinetics of subunit exchange, as monitored by depletion of homotetramers, is faster at 4°C than at 37°C (for rate constants and half-lives, see below). Hybrid tetramer formation between FT2 wild-type TTR and wild-type TTR was clearly observed after an 1-h incubation at 4°C. One half of the initial amount of homotetramers had disappeared after 12 h when incubated at 4°C, and after 40 h when incubated at 37°C. After 48 h, no further change in tetramer distribution was seen at 4°C, whereas equilibrium was reached only after 7 d at 37°C. The distribution of observed mixed tetramers was roughly symmetrical throughout the course of subunit exchange at both temperatures, with the 2:2 heterotetramer peak predominating.
Fig. 3.
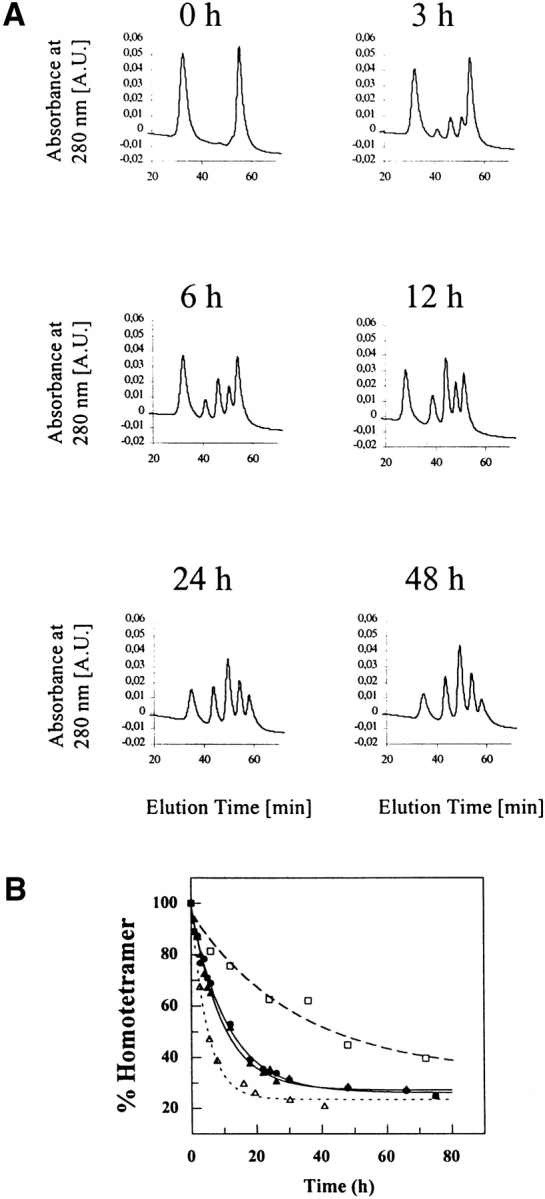
The time course of subunit exchange between FT2 wild-type TTR and wild-type TTR at 4°C, pH 7.4. (A) Formation of heterotetramers indicated by the appearance of three new peaks (peaks 2, 3, and 4) between the two initially present homotetramer peaks (peaks 1 and 5) as a function of time. (B). The rate of disappearance of FT2-wild-type TTR (filled triangles, solid line), wild type TTR (filled circles, solid line) homotetramers at 4°C (3.6 μM) and of wild-type TTR homotetramers at 37°C (3.6 μM; open squares, dashed line). An eightfold increase in TTR concentration (29 μM) at 4°C (open triangles, dotted line) leads to a twofold increase in exchange rate.
FT2 wild-type TTR exchanged subunits at a similar rate with V30M TTR at 4°C (Fig. 4A ▶). Equilibrium was achieved after 24 h at 4°C (Fig. 4C ▶). Subunit exchange was markedly slower at 37°C, with half of the initial V30M homotetramer being depleted after approximately 48 h. The exchange seemed to stagnate at this stage; even after 168 h (37°C), no further exchange could be detected. The V30M/FT2 wild-type TTR exchange at 37°C is not only significantly slower than wild-type/FT2 wild-type TTR exchange, but the extent of exchange is incomplete (compare Fig. 3B ▶ with Fig. 4C ▶). Within the first 12 h of mixing at 4°C, the distribution of heterotetramers was shifted toward tetramers having more FT2 wild-type subunits than V30M subunits (Fig. 4A ▶), likely because tetramers rich in wild-type subunits are more stable. This shift in tetramer population was more obvious at 37°C (Fig. 4B ▶).
Fig. 4.
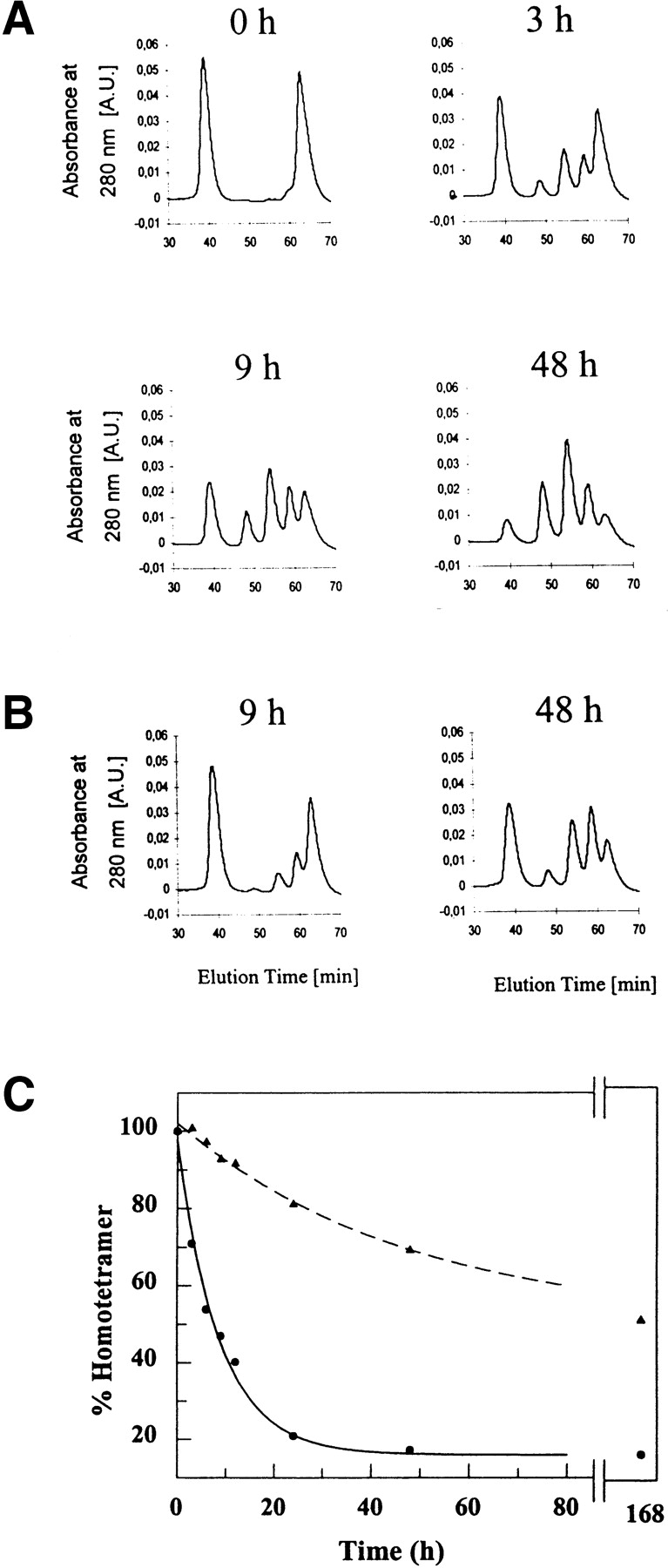
Ion-exchange chromatograms of V30M and FT2 wild-type TTR after incubation at 4°C (A) as a function of time and at 37°C (B) after 9 and 48 h. Heterotetramer formation between V30M and FT2 wild-type TTR at 4°C reaches equilibrium after 24 h (C). The tetramer distribution was shifted toward tetramers richer in FT2 wild-type TTR subunits within the first hours of incubation. At 37°C, this shift was more obvious (B). (C) Rate of disappearance of V30M homotetramer at 4°C (filled circles, solid line) and at 37°C (filled triangles, dashed line). The exchange rate is slow and incomplete even after 7 d of incubation at 37°C.
The formation of heterotetramers via simple mixing of homotetramers at pH 7 was dramatically inhibited in the presence of flufenamic acid. No heterotetramer peaks were formed after incubation for 1 w at 4°C (data not shown). Both flufenamic acid and thyroxine stabilize the TTR tetramer, apparently preventing the dissociation step that enables exchange.
Kinetics of subunit exchange
Figure 3B ▶ depicts the rate of subunit exchange as monitored by the decrease of homotetramer peaks in the ion exchange chromatogram as a function of time. The FT2 wild-type and wild-type homotetramer peaks showed similar rates of disappearance, indicating that addition of the FT2 did not alter the kinetics of subunit exchange appreciably (Fig. 3B ▶). The time course data fit to a first-order exponential function; the rate constant for disappearance of the wild-type TTR homotetramer peak at 4°C (kwt) was 0.089 ± 0.004 h−1, whereas the rate constant characterizing the disappearance of FT2 wild-type TTR homotetramers (kFT2-wt) was 0.105 ± 0.010 h−1, the corresponding half-lives of homotetramers being t½wt = 7.8 ± 0.4 h and t½FT2-wt = 6.6 ± 0.6 h, respectively. The data indicate that the rate of disappearance of the two homotetramers are similar. Similar kinetics are observed for subunit exchange between homotetrameric V30M TTR and FT2 wild-type TTR homotetramers at 4°C as indicated by the rate constant kV30M of 0.114 ± 0.013 h−1 and the corresponding half life t½V30M of 6.1 ± 0.6 h (Fig. 4 ▶ C). At 37°C, the rate of exchange decreased dramatically, and the rate constant for the exchange of wild-type TTR and FT2 wild-type TTR (kwt) was 0.030 ± 0.003 h−1 and of homotetrameric V30M TTR and FT2 wild-type TTR (kV30M) was 0.020 ± 0.002 h−1.
Discussion
Herein we introduce a new method to monitor subunit exchange in multimeric proteins using recombinant DNA technology combined with a chromatographic approach readily available in most laboratories. Importantly, the tandem Flag tag (FT2) does not appear to alter the thermodynamic stability, the kinetics of subunit exchange, or the amyloidogenicity of transthyretin, allowing careful studies on the formation and physical properties of mixed TTR tetramers, relevant to the heterozygous FAP diseases.
TTR is a remarkably stable protein under certain conditions. It does not thermally denature at temperatures <80°C; it requires very high concentrations of GdmCl for denaturation (Lai et al. 1997); and incubating the protein in 5% SDS in the absence of additional heating only dissociates the protein into dimers. The results presented in this paper are therefore somewhat surprising. We have been able to show conclusively for the first time that TTR undergoes subunit exchange under native conditions, showing that the tetramer dissociates on a biologically relevant timescale. We provide conclusive evidence that the native tetrameric structure is formed after subunit exchange, using equilibrium analytical ultracentrifugation data, and show that the thyroxine-binding sites are present in the mixed tetramers as discerned by the normal efficacy of thyroxine and flufenamic acid as inhibitors of transthyretin amyloid formation. The fact that subunit exchange is completely inhibited by the ligand flufenamic acid under physiological conditions indicates a dissociative mechanism for subunit exchange. At a minimum, the TTR tetramer has to disassemble into a trimer and a monomer, to form hybrid tetramers. Bernstein et al. (1970) suggested that dissociation into subunits might be the rate-limiting step of hybridization. Our data indicate that dissociation of the TTR tetramer occurs on a physiologically relevant timescale because subunit exchange between wild-type TTR can be observed after 1 h of incubation at 4°C, with equilibrium being reached in 1 d. In contrast, Quintas et al. (1999) suggested that dissociation of TTR tetramers at pH 7 resulted in formation of nonnative monomers that were unable to reform tetramers in experiments performed at very low TTR concentrations (0.3 μM). At physiological concentrations of TTR (3.6 μM at pH 7), we have not been able to detect monomeric TTR (Lashuel et al. 1998), and hence, our data show that the steady-state concentration of the TTR monomer is <1%, showing that reassociation of monomeric TTR is efficient at this protein concentration. Interestingly, we found that increasing the TTR concentration to 8 times physiological in plasma increased the rate of subunit exchange by a factor of two (rate constant, 0.208 ± 0.025 h−1; Fig. 3B ▶). Wild-type TTR and V30M TTR are expected to be mainly tetrameric under the conditions chosen in this study. Even if a small amount of monomer is present, the physiological conditions do not appear to induce a conformational change to the amyloidogenic intermediate; hence, the subunit can again participate in tetramer formation. The dissociation of tetramers appears to be the rate-limiting step of mixed tetramer formation, because the observed rates of hybrid formation correlate qualitatively with the amount of monomer present under physiological conditions or in physiological buffer at 4°C. The temperature dependence of subunit exchange indicates a decrease in stability of tetrameric TTR at lower temperatures. Chaotrope denaturation measurements clearly show decreased stability of TTR at lower temperatures (P. Hammarström and J.W. Kelly, unpubl.), suggesting a significant contribution from the hydrophobic effect to tetramer stability.
When commencing these studies, we wanted to see whether it was possible to see subunit exchange between an amyloidogenic tetramer (e.g., V30M TTR) and a less amyloidogenic tetramer such as wild-type TTR. From this study, we know that this occurs postsecretion, albeit slowly at 37°C. These studies introduce a new method to follow subunit exchange and provide the optimism that subunit exchange may afford a strategy for intervention in human amyloid disease. Such studies are underway in the laboratory.
Materials and methods
Plasmid construction
The human wild-type TTR gene was cloned into the NdeI-BamHI sites of the pET15b plasmid (Novagen), resulting in a vector containing the TTR gene with an N-terminally encoded (His)6-tag. PCR was used to amplify a region of this construct encoding the TTR gene, using a primer incorporating a Flag tag (Sigma-Aldrich) and NdeI site at the N-terminal end and another primer having an XhoI site at the C-terminal end. After digestion with appropriate enzymes, the PCR product was ligated to NdeI-XhoI–digested pET29a (Novagen) vector. The primers used for this PCR were (1) FlagNdeI sense primer: 5′-GGAATTCCATATGGAT TATAAGGATGACGATG-ACAAGGGTCCTACGGGCACC-3′ and (2) CHisXhoI anti-primer, 5′-CGGATCC-CTCCTCGAGTC CTTCCTTGGGATT-3′, where the underlined nucleotides in the FlagNdeI sense primer indicate the Flag tag. This procedure provided a vector encoding the TTR gene with an N-terminal Flag tag and a C-terminal (His)6-tag. Adding another (tandem) N-terminal Flag tag to this new construct was achieved by PCR amplification of the Flag-tagged TTR fragment with a second primer incorporating a Flag tag and NdeI site at the N terminus and another primer comprised of a stop codon between the TTR gene and an XhoI site at the C terminus. After digestion of the amplified fragment with NdeI and XhoI, it was ligated to NdeI-XhoI–digested pET29a vector. The primers used for this PCR were (1) FlagN2NdeI sense primer, 5′-GGAATTCCATATGGACTA CAAAGACGATGACGATAAGGATAT-AAGGATGACGAT-3′ and (2) D8FlagXhoI anti-sense primer, 5′-GGCGCCGCCGGC CTCGAGTCATTCCTTGGGATTGGTG-3′, where the underlined nucleotides in the FlagN2NdeI sense primer indicate the new Flag tag to be integrated. Different codons were used for the second Flag tag to prevent annealing to the Flag tag sequence already present in the template. The final construct encoded the 127 amino acids of the TTR gene product, preceded by a tandem Flag tag (FT2) with the sequence DYKDDDDKDYKDDDDK at its N terminus. DNA sequences were confirmed on both strands.
Expression and purification of TTR
Recombinant wild-type and V30M TTR were expressed in Escherichia coli BL21 (DE3; Stratagene) at 37°C. Selection was performed on Luria-Bertani (LB) broth supplemented, when appropriate, with 100 μg/mL ampicillin. Expression was induced by addition of 1 mM IPTG after cells reached an OD600 of 1.0. Cells were harvested 14 h after induction by centrifugation and were resuspended in 100 ml/L culture in 25 mM Tris-HCl (pH 8.0), 0.5 M NaCl and frozen at −80°C for 1h. Lysis was preformed by sonication on ice in the coldroom. The cell debris was pelleted by centrifugation. TTR was precipitated in 30% to 60% (w/v) ammonium sulfate and resuspended in 20 mL of 25 mM Tris-HCl (pH 8.0). The crude protein solution was desalted by FPLC in the same buffer using a HiPrep Desalt column (Pharmacia). Transthyretin was then loaded onto a Source 15Q anion exchange column (Pharmacia) and eluted (200 mL) during a 200- to 350-mM NaCl linear gradient in 25 mM Tris-HCl (pH 8.0), followed by size exclusion chromatography in 25 mM Tris-HCl (pH 7.4) using a Superdex 75 column (Pharmacia). Protein purity was assessed by SDS-PAGE, and composition was confirmed by electrospray ionization mass spectrometry (ESI-MS). FT2 wild-type TTR was expressed and purified as described above, with the following exceptions. Protein was precipitated in 28% to 56% (w/v) ammonium sulfate and eluted (400 mL) from the Source 15Q column using a 200 to 500 mM NaCl linear gradient.
Formation of heterotetramers
Purified homotetrameric TTR samples (3.6 μM) in 25 mM Tris-HCl (pH 7.4) were made from stock solutions by dilution with the same buffer. Equal concentrations of two different tetramers—FT2 wild-type TTR and wild-type TTR (or V30M TTR)—were then combined in an Eppendorf tube, mixed, and incubated at 4°C or 37°C. To investigate the concentration dependence of exchange, we also studied the exchange kinetics at a concentration of 29.5 μM tetrameric TTR at 4°C. To examine the effect of tetramer stabilizing small molecules such as flufenamic acid on heterotetramer formation, 3.6 μM TTR solutions were incubated with 7.2 μM flufenamic acid at 37°C for 30 min to allow binding. Flufenamic acid was added from a 432 μM stock solution in DMSO. After the preincubation, FT2 wild-type TTR and wild-type TTR with bound flufenamic acid were mixed in equal amounts and incubated at 4°C until analyzed.
Chromatographic analysis of heterotetramer formation
Separation of the tetramers was achieved by ion exchange chromatography using a smart system equipped with a μPeak UV monitor and a Mono Q PC 1.6/5 column (all Pharmacia). Above pH 7, the FT2 adds ∼6 negative charges to a TTR subunit; hence, the retention time of a tetramer is increased proportionally to the number of FT2-TTR subunits. To analyze heterotetramer formation, 40 μL of the mixed tetramer solution was combined with 10 μL of 25 mM Tris-HCl (pH 7.4) in a 100-μL Hamilton syringe and loaded onto the Mono Q column using a 50-μL sample loop. The column was washed for 8 min with 240 mM NaCl in 25 mM Tris-HCl (pH 8.0) at a flow rate of 25 μL/min. TTR was then eluted applying a 240- to 420-mM NaCl linear gradient in 25 mM Tris-HCl (pH 8.0) over 45 min at the same flow rate. The retention times of FT2 wild-type TTR and wild-type TTR homotetramers were established by combining 20 μL of each TTR solution (3.6 μM) with 10 μL of 25 mM Tris-HCl buffer (pH 7.4) in a 100-μL Hamilton syringe, which was then injected without preincubation. Integration of absorbance curves was accomplished using the smart manager 1.41 software according to the manufacturer's instructions. The rate of exchange was evaluated by the disappearance of homotetrameric peaks in the chromatogram and fitting the integrated surface area over time to a first-order kinetic equation. We were able to separate the five TTR heterotetramers resulting from subunit exchange on a preparative scale using the Source 15Q column described in the previous section. These peaks were collected and analyzed by reverse phase HPLC on a Waters 600E multisolvent delivery system coupled to a Waters 486 tunable absorbance detector (detection wavelength, 280 nm) using a C18 column (Vydac) to discern subunit composition. RP-HPLC analysis was performed using a linear increase in solvent B (95% CH3CN, 4.8% H2O, 0.2% TFA) relative to solvent A (95% H2O, 4.8% CH3CN, 0.2% TFA) at a flow rate of 1.0 mL/min. Masses of the eluted proteins were determined (ESI-MS).
Analytical ultracentrifugation
After complete exchange was confirmed by chromatographic analysis, the heteroterameric TTR mixture was analyzed by sedimentation equilibrium measurements. This was performed using a temperature-controlled Beckman Xl-I analytical ultracentrifuge (equipped with an An60Ti rotor and photoelectric scanner). A 100-μL TTR sample (7.2 μM) in 25 mM Tris-HCl (pH 7.4) was inserted in a double sector cell and analyzed at a rotor speed of 17000 rpm. When duplicate scans 3 h apart were superimposable, it ensured that equilibrium was reached (24 h). The data was analyzed using both the origin software from Beckman and nonlin (Johnson et al., 1981).
Protein stability determined by GdmSCN denaturation and fluorescence spectroscopy
Fluorescence emission spectra of FT2 wild-type TTR and wild-type TTR were recorded on an ATF 105 spectrofluorometer (Aviv) at 25°C using a 1-cm path length quartz cell. The excitation wavelength was 295 nm; emission was recorded over a spectrum from 310 to 410 nm. Data was presented as dependence of the ratio of the tryptophan fluorescence intensity at 355 and 335 nm. Native wild-type TTR showed a fluorescence maximum at 335 to 338 nm, whereas the unfolded protein showed maximum emission between 355 to 358 nm. We used the emission intensity ratio at 355/335 nm to follow denaturation by the exposure of the tryptophans. For denaturation studies, 1-μM (monomeric concentration) TTR solutions with GdmSCN concentrations ranging from 0 to 3 M were prepared in 50 mM phosphate buffer (pH 7.0) containing 1 mM EDTA and 1 mM DTT pH 7.0. The samples were incubated for 24 h before the measurement.
Fibril formation assay
Fibril formation assays on FT2 wild-type TTR were performed as described previously (Petrassi et al. 2000) and used Thioflavin T (ThT) fluorescence to quantify amyloid formation. This was performed to convince ourselves that the FT2 did not change the pH-dependent amyloidogenicity or change the binding of small molecule inhibitors. Four hundred ninty-five μL of a 7.2 μM TTR solution in 10 mM phosphate buffer, 100 mM KCl, 1 mM EDTA (pH 7.0) (5 μL of a 1.44 mM flufenamic acid solution in DMSO was added at this point in cases in which the small molecule inhibitor was desirable), incubated at 37°C for 30 min, was acidified by the addition of 500 μL of 200 mM acetate buffer, 100 mM KCl, 1 mM EDTA (pH 4.3) to yield a final pH of 4.4. The solutions were incubated at 37°C for 72 h without agitation. Fibril formation was then quantified by determining the turbidity of the solution at 400 nm after vortexing the sample for 5 sec. The extent of fibril formation of wild-type TTR in the absence of inhibitor was defined to be 100%.
For the ThT-binding assay, the samples were centrifuged for 10 min at 12 000g and the pellet was resuspended in 800 μL of 50 mM phosphate buffer (pH 7.0) containing 100 mM KCl, 1 mM EDTA, 1 mM DTT. Four μL of a 2mM solution of ThT was added, and the fluorescence of the sample was recorded over 450 to 600 nm after excitation at 440 nm. The emission intensity of ThT at 482 nm, characteristic of ThT bound to amyloid fibrils (Naiki et al. 1989), was recorded for the various samples.
Acknowledgments
We thank Songpon Deechongkit for assistance with analytical ultracentrifugation and mass spectrometry and Professor Voght for use of the SMART system. This work was supported by a grant from the National Institute of Health (DK46335-09) and by The Skaggs Institute of Chemical Biology and The Lita Annenberg Hazen Foundation. A postdoctoral fellowship to P.H. from the Wenner-Gren Foundations is gratefully acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.8901
References
- Bernstein, R.S., Robbins, J., and Rall, J.E. 1970. Polymorphism of monkey thyroxin-binding prealbumin (TBPA): Mode of inheritance and hybridization. Endocrinology 86 383–390. [DOI] [PubMed] [Google Scholar]
- Colon, W. and Kelly, J.W. 1992. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 31 8654–8660. [DOI] [PubMed] [Google Scholar]
- Holmgren, G., Ericzon, B.-G., Groth, C.-G., Steen, L., Suhr, O., Andersen, O., Wallin, B.G., Seymour, A., Richardson, S., Hawkins, P.N., et al. 1993. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 341 1113–1116. [DOI] [PubMed] [Google Scholar]
- Holmgren, G., Haettner, E., and Lindström, A. 1988. Homozygosity for the transthyretin-met30-gene in two Swedish sibs with familial amyloidotic polyneuropathy. Clin. Genet. 34 333–338. [DOI] [PubMed] [Google Scholar]
- Johnson, M.L., Correia, J.J., Yphantis, D.A., and Halvorson, H.R. 1981. Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques. Biophys. J. 36 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno, K., Palha, J.A., Miyakawa, K., Saraiva, M.J.M., Ito, S., Mabuchi, T., Blaner, W.S., Iijima, H., Tsukahara, S., Episkopou, V., et al. 1997. Analysis of amyloid deposition in a transgenic mouse model of homozygous familial amyloidotic polyneuropathy. Am. J. Pathol. 150 1497–1508. [PMC free article] [PubMed] [Google Scholar]
- Lai, Z., Colon, W., and Kelly, J.W. 1996. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 35 6470–6482. [DOI] [PubMed] [Google Scholar]
- Lai, Z., McCulloch, J., Lashuel, H.A., and Kelly, J.W. 1997. Guanidine hydrochloride-induced denaturation and refolding of transthyretin exhibits a marked hysteresis: Equilibria with high kinetic barriers. Biochemistry 36 10230–10239. [DOI] [PubMed] [Google Scholar]
- Lashuel, H.A., Lai, Z., and Kelly, J.W. 1998. Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: Implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry 37 17851–17864. [DOI] [PubMed] [Google Scholar]
- Liu, K., Cho, H.S., Lashuel, H.A., Kelly, J.W., and Wemmer, D.E. 2000. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat. Struct. Biol. 7 754–757. [DOI] [PubMed] [Google Scholar]
- Naiki, H., Higuchi, K., Hosokawa, M., and Takeda, T. 1989. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal. Biochem. 177 244–249. [DOI] [PubMed] [Google Scholar]
- Petrassi, H.M., Klabunde, T., Sacchettini, J., and Kelly, J.W. 2000. Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. J. Am. Chem. Soc. 122 2178–2192. [Google Scholar]
- Quintas, A., Saraiva, M.J.M., and Brito, R.M.M. 1999. The tetrameric protein transthyretin dissociates to a non-native monomer in solution: A novel model for amyloidogenesis. J. Biol. Chem. 274 32943–32949. [DOI] [PubMed] [Google Scholar]
- Saraiva, M.J.M. 1995. Transthyretin mutations in health and disease. Hum. Mutat. 5 191–196. [DOI] [PubMed] [Google Scholar]