

Abstract
Maltose binding protein (MBP) is widely used as a model for protein folding and export studies. We show here that macroscopic aggregates form transiently during the refolding of MBP at micromolar protein concentrations. Disaggregation occurs spontaneously without any aid, and the refolded material has structure and activity identical to those of the native, nondenatured protein. A considerable fraction of protein undergoing folding partitions into the aggregate phase and can be manually separated from the soluble phase by centrifugation. The separated MBP precipitate can be resolubilized and yields active, refolded protein. This demonstrates that both the soluble and aggregate phases contribute to the final yield of refolded protein. SecB, the cognate Escherichia coli cytosolic chaperone in vivo for MBP, reduces but does not entirely prevent aggregation, whereas GroEL and a variety of other control proteins have no effect. Kinetic studies using a variety of spectroscopic probes show that aggregation occurs through a collapsed intermediate with some secondary structure. The aggregate formed during refolding can convert directly to a near native state without going through the unfolded state. Further, optical and electron microscopic studies indicate that the MBP precipitate is not an amyloid.
Keywords: Aggregation, intermediates, folding, MBP
Aggregation and precipitation are widespread problems encountered during the folding of polypeptide chains, both in vivo and in vitro. Protein aggregates are found associated with several human diseases (Carrell and Lomas 1997; Harrison et al. 1999), and precipitation of overexpressed proteins is a major problem often encountered in the biotechnology industry. Folding of nascent polypeptides within cells normally occurs at high protein concentrations, of greater than 100 mg ml−1. Under such conditions, protein aggregation is a significant problem. Cells overcome this challenge by producing a set of proteins termed molecular chaperones (Morimoto et al. 1994) which are characterized by their ability to bind and sequester unfolded polypeptide chains, thus preventing their aggregation. Detailed study of the aggregation process has been difficult due to the irreversible nature of most protein aggregation reactions. Although it is reversible, transient aggregation during refolding has been observed in few cases (Pecorari et al. 1996; Silow and Oliveberg 1997; Silow et al. 1999); the size of the aggregates is generally small, and no visible precipitation occurs therein. Aggregation involving protein folding intermediates (Brems 1990; King et al. 1996; Pecorari et al. 1996; Raman et al. 1996) and aggregation from the unfolded state (Silow and Oliveberg 1997; Silow et al. 1999) of proteins have been reported. Aggregation is thought to be a nonproductive, off-pathway reaction which competes with correct folding reactions (Kiefhaber et al. 1991).
Maltose binding protein (MBP) is a 370 amino acid, two-domain protein localized in the periplasm of E. coli. The folding of MBP has been studied, but most of the previous investigations were carried out using low protein concentrations, in the submicromolar range (Chun et al. 1993).We show here for the first time that MBP refolding is aggregation-prone, even at concentrations as low as a few micromolar. However, the large-scale aggregation and precipitation of refolding MBP is completely and spontaneously reversible, without any external aid. The redissolved protein is recovered in an active, folded form. We also examined the kinetics of MBP folding under nonaggregating conditions, using several spectroscopic probes, utilizing both rapid mixing stopped-flow and manual mixing techniques. We conducted these studies to identify intermediates that might be involved in the aggregation observed at higher protein concentrations. In addition, the effect of the cognate E. coli chaperone, SecB, on the aggregation of MBP was characterized.
Results
Folding of MBP under nonaggregating conditions
The refolding kinetics of MBP were examined using two different probes, far-UV circular dichroism (CD) and intrinsic tryptophan fluorescence, using low protein concentrations, below 0.7 μM, where no visible aggregation of protein during folding is observed (Fig. 1 ▶). The results show that the folding of MBP occurs in multiple kinetic phases, each phase representing the formation of at least one kinetic intermediate or of native protein.
Fig. 1.

Kinetics of refolding of MBP at 25°C in the absence of aggregation at low protein concentrations. Refolding was monitored by the changes in intrinsic tryptophan (Trp) fluorescence (a) and changes in circular dichroism (CD) mean residue ellipticity (M.R.E.) at 222 nm (b) that occur with time after dilution of denaturant. The fluorescence measurements in (a) were made in a stopped-flow machine after rapid mixing (deadtime = 1.4 msec) at a protein concentration of 0.5 μM. Each curve is an average of at least a dozen independent traces. Curves in (a) were referenced with respect to the unfolded state signal which is set to zero. The CD measurements in (b) were made following manual mixing of solutions. The MBP concentration was 0.7 μM. The broken line in (b) represents the residual ellipticity in unfolded MBP. In both panels, dots represent the data points and the solid lines represent exponential fits to the data.
The increase in intrinsic tryptophan fluorescence that accompanies folding occurred in three kinetic phases (Fig. 1a ▶). A large, burst phase change occurring in the deadtime of mixing was followed by observable changes that occurred in two well separated time domains, with apparent rate constants of 0.50 ± 0.02 and 0.041 ± .002 s−1, respectively.
To determine the secondary structural content of the product(s) of the burst and fast phases, which are complete by 10 sec, we made manual measurements of ellipticity at 222 nm. Figure 1b ▶ shows that the mean residue ellipticity at 222 nm increased from 10 sec (the earliest time point of measurement) to 100 sec in a single exponential process with an apparent rate constant of 0.058 ± .017 sec−1. Thus, within experimental error, the rate constants obtained for the slow phase using tryptophan (Trp) fluorescence and CD are identical. Extrapolation of the single exponential process to t = 0 indicates that only 30% of native-state ellipticity signal is recovered by 10 sec. Since stopped-flow CD measurements were not possible, it is not possible to determine what fraction of the native-state ellipticity signal is recovered in the burst phase of folding. The results in Figure 1b ▶ do, however, indicate that the products of the burst and fast phases of folding represent only 30% of the secondary structure content of N. The kinetic parameters of all phases measurable by manual mixing at 25°C were found to be independent of protein concentration in the range 0.05 to 0.7 μM MBP (data not shown), suggesting that no concentration-dependent process such as aggregation occurs under these conditions at these low concentrations.
Because MBP is a large protein with several Pro residues, the folding pathway is expected to be complex and is likely to involve several intermediates and parallel pathways of folding. It should be emphasized that the objective of the present study was not to obtain a detailed and complete picture of MBP folding. Rather, it was to obtain some insight into the structural features of intermediate(s) that might be involved in the aggregation process that occurs when folding is carried out at higher protein concentrations (> 2 μM).
Spontaneously reversible precipitation during MBP refolding
When MBP unfolded in guanidine hydrochloride (GdnHCl) at high concentrations was diluted into refolding buffer, aggregation and large-scale precipitation perceptible to the naked eye occurred (Fig. 2a–d ▶). Aggregation was also detected by optical scatter measurements in a photometer (Fig. 2e ▶). The inset in Figure 2e ▶ shows that at a final MBP concentration of 28 μM, there was a lag of about 2 sec between denaturant dilution and the initiation of aggregation, and that aggregation was highly cooperative. Aggregation was maximal after about 60 sec and then gradually decreased. Refolding studies were also carried out at final MBP concentrations of 17, 34, and 51 μM. The lag phase was present at all four protein concentrations. This suggests that an intermediate on the folding pathway is likely to be responsible for the aggregation. It is also possible that the lag phase represents a slow nucleation event. The timescale of the lag phase indicates that even such a nucleation event is likely to involve intermediate(s) formed on the second timescale rather than the unfolded state. Refolding experiments at final MBP concentrations of 2, 4, and 8 μM were also carried out using manual mixing. Aggregation was monitored by following the intensity of scattered light at 320 nm as a function of time (Fig. 2f ▶). As expected for an aggregation process, the kinetics were concentration-dependent and the extent of aggregation increased with the increase in protein concentration.
Fig. 2.




Precipitation and spontaneous resolubilization of MBP while refolding in GdnHCl at high protein concentrations. (a–d) zero time (before addition of MBP), and after 30 sec, 2 min, and 12 hr equilibration of refolding 25 μM MBP in 0.1 M GdnHCl. Sample was mixed intermittently by inverting the cuvette. (e and inset) Aggregation kinetics of 28 μM MBP following rapid mixing in a stopped-flow device, of unfolded MBP with refolding buffer containing 0.4 M GdnHCl. (f) Aggregation kinetics of 2 (dashed/dotted line), 4 (dashed line) and 8 (solid line) μM MBP in 0.2 M GdnHCl following manual mixing. (g) Near-UV CD spectra of native MBP (dashed line) and that of the precipitated and spontaneously redissolved, refolded protein (dotted line). All measurements were made at pH 7.3 and 24°C.
The refolded protein obtained after spontaneous redissolution of the precipitate is similar to the native form, as deduced from near-UV CD spectra (Fig. 2g ▶), far-UV CD, native gel electrophoretic mobility, and fluorescence spectroscopy (data not shown). The affinity of spontaneously resolubilized and refolded protein for maltose was measured by fluorimetric titration and was found to be 1.1 μM, identical to the published value for native protein (Ganesh et al. 1997). In another set of experiments, the binding affinity of maltose to MBP present in the soluble supernatant, and to MBP obtained upon manual resolubilization of the separated pellet were also found to be identical to that of native protein.
Quantification of the partitioning of refolding MBP molecules between the soluble supernatant and the pellet, done using absorbance and fluorescence spectroscopy, indicates that roughly half the total protein is aggregated after a minute of refolding. This fraction declines gradually to zero at longer times of refolding (Table 1). Thus, folded and active protein arises from both the soluble and aggregate phases. The results of the absorbance studies were in qualitative agreement with those of scattering experiments, suggesting that scattering could be used to follow the progress of aggregation and disaggregation. The time taken for the intensity of light scattering at 320 nm to fall to 50% of the maximal value (t50%) has been used as a convenient index to analyze refolding from the aggregated state.
Table 1.
Mass balance for the aggregation and disaggregation reactions
| Time (s) | A280 (supernatant) | A280 (resuspended pellet) | A280 (supernatant) + A280 (pellet) | % in pellet |
| 20 | 0.255 | 0.060 | 0.315 | 19 |
| 45 | 0.201 | 0.100 | 0.301 | 33 |
| 60 | 0.189 | 0.140 | 0.329 | 43 |
| 90 | 0.193 | 0.122 | 0.315 | 39 |
| 120 | 0.208 | 0.128 | 0.336 | 38 |
| 180 | 0.195 | 0.108 | 0.303 | 36 |
| 360 | 0.267 | 0.043 | 0.310 | 14 |
| 3600 | 0.305 | 0.004 | 0.309 | 1 |
Relative amount of maltose-binding protein (MBP) present in the macroscopic aggregate as a function of time of refolding. Five μM MBP was refolded in 1 ml buffer at 22°C. At various times, the sample was spun down. The supernatant was removed and the pellet resuspended in 1 ml of fresh refolding buffer. The absorbance at 280 nm was measured in both supernatant and pellet. The final expected A280 based on the known extinction coefficient of MBP is 0.301.
Disaggregation of MBP during refolding
The disaggregation of the MBP precipitate was further characterized by separating the aggregate from soluble protein by a brief centrifugation step, as described in Materials and Methods. The kinetics of refolding of protein in the soluble (nonaggregated) fraction were monitored (upper trace in Fig. 3a ▶). The nonaggregated protein appears to fold with a rate constant of .01 sec−1, similar to that observed for the slow phase of folding at low protein concentrations (Fig. 1 ▶), although this rate is probably an underestimate because a large fraction of the folding reaction was lost in the first 100 sec that could not be observed. The aggregated protein was rapidly solubilized by the addition of fresh buffer. The kinetics of refolding in the resolubilized aggregate thus obtained were monitored by Trp fluorescence. As can be seen from the lower trace in Figure 3a ▶, the fluorescence of the resolubilized aggregate did not change with time; that is, the aggregate reached a native-like state within the deadtime of the resolubilization process.
Fig. 3.

Kinetics of refolding of aggregated and nonaggregated protein. (a) Refolding of unaggregated protein present in supernatant (top trace) and protein from the resolubilized pellet (bottom trace), after centrifugation, were measured at 25°C as described in Materials and Methods. A single exponential fit of the data obtained from the supernatant yields an observed rate constant of 0.01 sec−1, while the Trp fluorescence in the resolubilized pellet does not change appreciably with time. (b) Refolding of unaggregated protein present in the supernatant upon refolding at 4°C, using three different protein concentrations: 2 μM (solid line), 10 μM (inverted triangles) and 20 μM (circles). In the latter two cases, aggregated protein was removed by a brief centrifugation prior to measurement. The centrifugation step was omitted for the 2 μM MBP sample because no aggregation was detected for that concentration at the temperature of measurement. In each case, the MBP was refolded in 0.26 M GdnHCl for 10 sec at 4°C prior to removal of aggregate by centrifugation. Refolding was monitored by measurement of tryptophan fluorescence as a function of time of folding. In each case, fluorescence values at any time of folding were normalized to a value of 1 for the value of fluorescence after 500 sec of folding.
At high protein concentrations, folding and aggregation occur in parallel, whereas at low protein concentrations, no aggregation occurs. To compare folding rates under these two conditions, we carried out folding experiments at 4°C using different protein concentrations. At 4°C, aggregation occurred to a lesser extent than at 25°C, and at a protein concentration of 2 μM, aggregation did not occur at the lower temperature. As described above, at higher protein concentrations, aggregated protein was separated from soluble protein by a brief centrifugation. Since folding is considerably slower at 4°C than at 25°C, only a small extent of folding occurred in the time taken to remove the aggregate by centrifugation. Figure 3b ▶ shows that the slow phase of folding at high protein concentrations where aggregation was seen occurred at the same rate, and had the same amplitude, as that of the slow phase of folding at low protein concentrations where no aggregation was seen. The figure shows that even when aggregation occurred, the protein that was not aggregated folded with kinetics identical to those observed at low concentrations in the absence of aggregation.
Effect of chaperones on MBP aggregation and disaggregation
SecB is an E. coli chaperone (Kumamoto and Beckwith 1983) that binds MBP and a subset of proteins destined for export to the periplasm, and maintains them in an unfolded translocation competent state (Weiss et al. 1988). In our experiments, the addition of SecB to the refolding buffer led to a decrease in t50%, whereas another chaperone, GroEL (± GroES, ATP) had little or no effect on t50% (Fig. 4 ▶). Other control proteins such as RNase A, lysozyme, and BSA had no effect (data not shown). The t50% value initially decreased with an increase in SecB concentration, but reached a plateau value of about 80 sec (Fig. 4b ▶). Thus, high concentrations of SecB diminish but cannot prevent MBP aggregation. SecB decreased t50% even when added after the onset of aggregation (Fig. 4a ▶, inset) and at times when there was no unfolded protein in the soluble supernatant. The absence of unfolded protein in the soluble supernatant at these times can be ascertained from the fact that the fluorescence in the supernatant does not change with the time of folding. SecB had no effect on the duration of the lag phase of aggregation (data not shown).
Fig. 4.
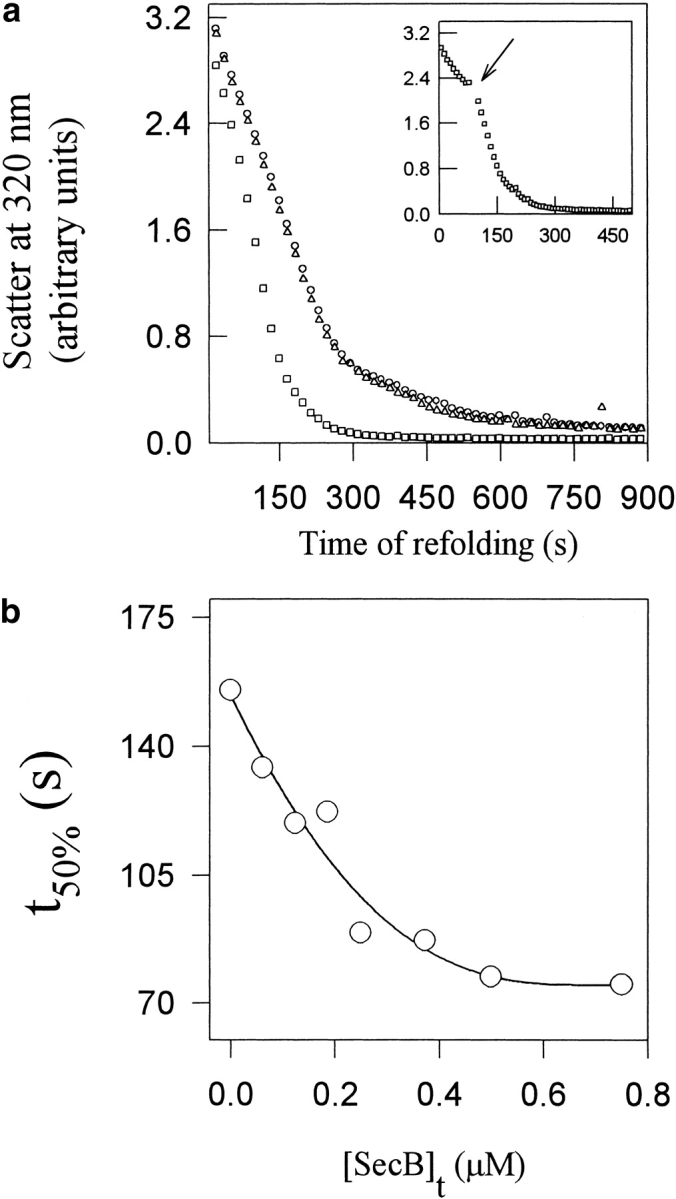
Specific effect of SecB on MBP aggregation. (a) Disaggregation of 2 μM MBP refolding in the absence (circles) and presence of 1 μM SecB (squares) or 1 μM GroEL (triangles). The inset shows the effect of pulsed addition of 1 μM SecB at the indicated time arrow). (b) Effect of SecB concentration on the disaggregation kinetics of 1.4 μM MBP. In all cases, the temperature was 30°C and the samples were stirred magnetically.
Effect of temperature on MBP aggregation
To gain insight into the driving forces for MBP aggregation, we carried out studies of MBP aggregation as a function of temperature. Although the rate of disaggregation was faster at higher temperatures (Fig. 5 ▶), the extent of MBP aggregation was enhanced in a cooperative manner with increasing temperature (Fig. 5 ▶, inset).
Fig. 5.

MBP aggregation as a function of temperature. Aggregation kinetics were monitored by optical scatter at 320 nm, during the refolding of 2 μM MBP in refolding buffer at (○) 24°C, (□) 31°C, (▵) 35°C and (▽) 43°C. The inset shows the extent of aggregation after 10 sec of refolding at various temperatures.
The MBP precipitate is not an amyloid
The nature of the MBP precipitate was examined using electron microscopy and Congo red dye binding. As a positive control, insulin, which is known to form amyloid fibrils under appropriate conditions (Kedar et al. 1976), was also examined. Unbranched, elongated amyloid fibrils forming a dense meshlike network were observed clearly in the case of the insulin precipitate (Fig. 6a ▶), whereas the MBP precipitates were smaller and had a granular appearance (Fig. 6b ▶). In the Congo red dye binding assay (data not shown), the bound dye exhibited blue-green birefringence only in the case of insulin (Kedar et al. 1976), not with the MBP precipitate. The resolution of the electron microscopy was insufficient to permit detailed characterization of the MBP precipitate.
Fig. 6.

Electron micrographs of (a) insulin amyloid fibrils and (b) the transient MBP precipitate obtained during refolding. Scale bar, 200 nm.
Discussion
Folding reactions of MBP
The data in Figure 1 ▶ suggest that in less than a few milliseconds of refolding at low protein concentrations, the unfolded state collapses to a compact state (Ib). MBP has eight tryptophan residues distributed between the two domains in the molecule. The large, burst phase Trp fluorescence change accompanying the formation of Ib is therefore likely to reflect a global event. Although it was suggested earlier (Chun et al. 1993) that an initial collapse of MBP into a folding intermediate could occur within milliseconds of denaturant dilution, followed by other slow folding reactions, the present study is the first to examine the folding of MBP with millisecond time resolution.
In the next few seconds of folding, we observed minor changes in the burial of Trp residues. At the end of a few seconds of refolding, MBP has 30% of the native secondary structure, and there is substantial burial of Trp residues. We denote the collection of intermediates present after 2 seconds of refolding as Is. The major slow folding reaction of MBP occurs with an apparent half-life of about 15 sec (monitored by CD and Trp fluorescence). During this period, the protein acquires native secondary structure and tertiary structure. We denote the species formed at the end of this phase as N. The slow folding reaction is not due to proline isomerization (Ganesh et al. 1999). This was established by double-jump studies, which showed that the observed rate for the slow-phase amplitude buildup was about 100-fold faster than that expected for Pro isomerization (Ganesh et al. 1999).
N has native-like secondary structure and tryptophan fluorescence, and is an active form of MBP capable of binding maltose. Binding of maltose to MBP leads to a 15% decrease in Trp fluorescence. In refolding experiments carried out in the presence of maltose, we observed maltose binding only during the later stages of the slow phase, after about 3 min of refolding (data not shown). The inclusion of maltose had only marginal effects on the kinetics of the slow phase and did not affect the aggregation observed at high concentrations of MBP (data not shown).
The folding kinetics of MBP were studied here at low protein concentrations where not only is visible aggregation absent, but any events involving soluble aggregates must also be absent, because folding is concentration-independent. The folding kinetics were not studied in exhaustive detail because our goal was not to define the folding mechanism of MBP, but rather to determine which folding reaction occurs in the same time domain as the aggregation process. For this purpose, it is of course important to demonstrate that the kinetics of the folding reaction occurring along with the aggregation reaction are independent of protein concentration, as seen in Figure 3b ▶. Characterization of this folding reaction will allow determination of the structural features of the intermediate that are likely to be responsible for the aggregation process.
Folding and aggregation of MBP
The folding of MBP at micromolar concentrations involves the reversible formation of macroscopic aggregates. Since there is a lag phase in the formation of aggregates during folding, it is unlikely that aggregation is a direct result of a transfer of unfolded protein to refolding conditions. The formation of any aggregate, whether soluble or insoluble, would be expected to be strongly dependent on the concentration of protein. The lack of concentration dependence of the lag phase suggests that aggregation occurs directly from a folding intermediate that populates the folding pathway. It is also possible that the lag phase represents a nucleation event and the apparent lack of concentration dependence is simply because the size of the nucleus is small and the concentration range examined was insufficient. In either case, because of the timescales involved, aggregation is unlikely to be occurring directly from the unfolded state. Aggregates appear to be formed on the timescale of seconds and are completely dissociated after several minutes. However, the observed dissociation is likely to involve the net effect of both disaggregation and reaggregation events. It is difficult to characterize aggregation processes on the timescale of seconds. Hence, we attempted to first characterize the folding pathway of MBP at low concentrations where no aggregation occurs. On the timescale of a few seconds associated with aggregation, the intermediate(s) formed are collapsed molecules with about 30% of native secondary structure.
That we were able to observe only up to about half the amount of total protein in the aggregated state could be due to the following reasons. MBP has 21 proline residues, and during folding the proline residues will be in a mixture of cis and trans conformations. It is quite likely that only a subset of these conformational isomers are able to participate in the aggregation process. Secondly, the changes in scatter as a function of time illustrated in Figure 2 ▶ reflect the net effects of aggregation and disaggregation. It is not possible to deduce the rates for either aggregation or disaggregation from the data. The change in scatter with time shown in Figure 2f ▶ is clearly and strongly concentration-dependent, as expected. Kinetic partitioning is likely to occur between the aggregation and the direct folding reaction to N, and would determine the fraction of molecules that fold through the aggregation pathway instead of folding directly.
Characterization of the disaggregation process
It is particularly interesting that when the aggregate is resolubilized, the resolubilized protein does not exhibit any change in fluorescence in a slow phase that is typically observed during MBP folding (Fig. 3a ▶, bottom curve). If the aggregate were to unfold to Is or earlier intermediates before refolding, a time-dependent change in Trp fluorescence that characterizes the Is→N transition would have been detected, as is the case for the protein in the soluble supernatant (Fig. 3a, ▶top curve). Thus, the aggregate appears competent to fold directly to N without first unfolding to U or even to Is. Hence the aggregated state appears to be an on-pathway aggregated intermediate.
Nature of the aggregation process
For reasons that are presently unclear, the temperature dependence of aggregation is surprisingly cooperative. One possible inference is that the aggregation process involves the participation of temperature-dependent hydrophobic forces, involving burial of nonpolar surfaces between protein molecules (Xie and Wetlaufer 1996). It is also possible that the observed effects are due to differential stabilization of an aggregation-prone intermediate with respect to the native state, as a function of temperature. Since MBP is a two-domain protein, one attractive possibility is that the precipitate consists of domain-swapped oligomers of the protein (Schlunegger et al. 1997). This would explain the observation that the precipitate is formed from a late intermediate, and that it can convert directly to the native state without unfolding.
Wild-type MBP can be expressed to levels as high as100 mg/L of E. coli culture. The protein can be produced either in the cytoplasm or in the periplasm. The in vivo levels of MBP in the periplasm can be as high as 1 mM (Dietzel et al. 1978). All of the protein is found in the soluble form. However, several MBP mutants form inclusion bodies in vivo and insoluble aggregates in vitro (Betton et al. 1998; Raffy et al. 1998). Studies of the folding pathways of such mutants and examination of those precipitates may yield further insights into the structural features responsible for reversible aggregation of the wild-type protein.
Role of cellular chaperones
Nascent polypeptide chains destined for translocation are expected to undergo competition between folding, misfolding events like aggregation, and the actual process of productive export. Folded or aggregated MBP is translocation-incompetent, and the cytosolic chaperone SecB is required for the efficient translocation of MBP (Collier et al. 1988). SecB is thought to function by maintaining a subset of precursor polypeptides in an export-competent form by preventing aggregation or folding, and delivering the precursors to the export apparatus located in the bacterial inner membrane (Hardy and Randall 1993). It has been shown that SecB can prevent the irreversible aggregation of two of its natural substrates, OmpA and PhoE (Lecker et al. 1990; Breukink et al. 1992). While we cannot rule out a role for SecB in prevention of MBP aggregation during refolding, the data also suggest that SecB promotes the rapid disaggregation of MBP without greatly enhancing the final yield of refolded MBP.
It was shown previously that the chaperone GroEL can interact with proOmpA and prePhoE (Lecker et al. 1989), two proteins destined for export across the bacterial membrane. The observation that SecB but not GroEL/GroES or other proteins specifically promotes MBP disaggregation is also of interest. The effect of GroEL on the refolding of MBP at low protein concentrations has been described (Sparrer et al. 1996) and was reproduced during the course of the present work (data not shown). However, our present findings indicate that the GroE chaperone system is incapable of preventing MBP aggregation at physiologically relevant concentrations.
Although the precursor form of MBP is the in vivo substrate for SecB in the E. coli cytoplasm, SecB binding sites are located primarily in the mature sequence of MBP (Gannon et al. 1989); therefore, the present findings using mature MBP are also relevant to the in vivo folding situation. It has been proposed (Betton and Hofnung 1996) that a newly exported protein in the bacterial periplasm undergoes kinetic competition among folding, proteolytic degradation, and aggregation. Mature MBP is located primarily in the bacterial periplasm, and thus it is important to determine whether there are any periplasmic chaperones which may suppress MBP aggregation (Wulfing and Pluckthun 1994; Betton and Hofnung 1996). Most known periplasmic chaperones are involved in either Pro isomerization or disulfide bond formation (Missiakas and Raina 1997). Neither of these processes are involved in the aggregation reported here. Initial experiments on the addition of bacterial periplasmic extracts to refolding MBP did not reveal any appreciable effect on the aggregation process (data not shown).
General implications and conclusion
MBP is widely used as a fusion partner in high-level recombinant protein expression and purification (di Guan et al. 1988), and commercial vectors are readily available for that purpose. This is due in part to the high expression levels of soluble MBP achievable in E.coli. A recent report describes the efficient recovery of different proteins fused to MBP in a soluble form (Kapust and Waugh 1999). This high yield of soluble recombinant proteins may be related to the unusual feature of MBP folding described here. The finding that spontaneous refolding can occur from a macroscopic aggregate in the case of MBP raises the intriguing possibility that such refolding reactions may occur in vivo as well as during the folding of other proteins.
A recent set of detailed investigations on the folding of interleukin 1β (IL-1β) (Finke et al. 2000a, b) has shed more light on the critical late stage at which commitment of a folding polypeptide to either the native state or insoluble aggregates occurs. A nucleation-dependent irreversible aggregate formation from the unstructured (unfolded) ensemble describes the off-pathway, nonproductive processes in the case of IL-1β. The formation of self-solubilizing macroscopic precipitates and aggregation through folding intermediates in the case of MBP thus represent a different class of protein aggregation reaction. Further studies on the aggregation of several other proteins would prove vital in unraveling this complex process.
The classic studies by Anfinsen and colleagues (Anfinsen 1973) first showed that the sequence of a protein can contain sufficient information to specify its three-dimensional structure. It has been suggested that in addition, other molecules such as molecular chaperones (Morimoto et al. 1994) may be required for protein folding at high concentrations that occur in vivo. The present work shows that in the case of MBP, no additional information besides the amino acid sequence is required to attain the correct fold, and that correct and complete folding occurs even after large-scale protein aggregation and precipitation.
Materials and methods
Materials and protein purification
All reagents were from Sigma Chemical Co. MBP and SecB were purified as described earlier (Ganesh et al. 1997; Panse et al. 1998). BSA, lysozyme and RNase A were from Sigma and used without further purification. All solutions were filtered through a 0.22 μm filter. Stock protein and other working solutions were centrifuged before use to remove any insoluble matter.
Aggregation assay
Aggregation during MBP refolding was followed by monitoring the changes in light scatter intensity at 320 nm in a JASCO 7850 spectrophotometer, a JASCO FP 777 spectrofluorimeter, a SPEX Fluorolog fluorimeter, or a BioLogic SFM4 stopped-flow instrument with a deadtime of 1.4 msec. Typically, 200 μM of unfolded MBP in 3 M GdnHCl was diluted to the desired final concentration in refolding buffer, and aggregation was monitored as a function of time. At 24°C, aggregation was observed for final concentrations of MBP greater than 2 μM and at final GdnHCl concentrations in the range of 0.05–0.4 M. Manual mixing aggregation assays were performed at different temperatures and in the presence of several additives.
Spectroscopic methods
Far-UV and near-UV CD spectra were acquired in a JASCO 500A spectropolarimeter using a 1mm (far-UV) or 5 mm (near-UV and refolding kinetics at 222 nm) quartz cuvette. MBP concentrations of 0.7 μM, 2 μM and 20 μM were used for the refolding (monitored at 222 nm), far-UV, and near-UV experiments, respectively. The deadtime for the manual mixing CD refolding experiment was 4 sec. Steady-state fluorescence emission spectra and refolding kinetics traces (excitation at 280 nm and emission at 341 nm) after manual mixing were recorded in a JASCO FP777 spectrofluorimeter. All experiments were conducted in 10 mM HEPES buffer containing 150 mM NaCl (pH 7.3) (refolding buffer). For equilibrium near-UV CD measurements, 71 μL of 280 μM unfolded MBP (in 3 M GdnHCl) was mixed with 930 μL refolding buffer (final MBP concentration during refolding was 20 μM). Precipitation visible to the naked eye occurred but faded away completely with time, and CD spectra were recorded after equilibration as described (Ganesh et al. 1997).
Kinetics experiments and data analyses
The kinetics of MBP refolding under nonaggregating conditions were monitored at a protein concentration of about 0.5 μM MBP in refolding buffer containing 0.15 M GdnHCl, at 24°C. Rapid mixing experiments were performed in a Biologic SFM 4 stopped-flow instrument. The excitation wavelength was set to 280 nm, and correspondingly a 320 nm Oriel cut-off filter was used to monitor Trp fluorescence. Changes in signal intensity as a function of time in all kinetics experiments were fit to either of the equations aα+Σai*exp(-ki*t) or a0+Σai*[1-exp(-ki*t)], where ai represents the amplitude change associated with the process occurring with an observed rate constant of ki. a0 is the amplitude change occurring within the burst phase and aα represents the amplitude achieved at equilibrium. All curve-fitting procedures were performed as described (Agashe et al. 1995).
Refolding kinetics from the aggregated state
Experiments were also performed to follow the refolding from the precipitate. Twenty μM MBP was first refolded in 0.1 mL of 0.35 M GdnHCl for 20 sec at 30°C. To separate the precipitate from soluble protein, the sample was then centrifuged at 12,000 × g for 10 sec. All of the supernatant (0.1 mL) was removed and diluted to 1 mL in refolding buffer. The folding kinetics of MBP in the diluted supernatant were monitored by Trp fluorescence at 340 nm. The pellet which contained the aggregated MBP was immediately redissolved in 1 mL of fresh refolding buffer, briefly recentrifuged for 10 sec and then transferred to a 1-mL fluorescence cuvette. The increase in Trp fluorescence intensity of the material in the redissolved pellet was monitored at 340 nm as a function of time.
Mass balance and activity of recovered MBP
In a refolding experiment carried out at a final MBP concentration of 20 μM, the sample was centrifuged after 20 sec of refolding. The supernatant was removed from the pellet and the latter was separately resolubilized with fresh refolding buffer. The binding affinities of maltose to MBP present in the soluble supernatant and to MBP obtained from the resolubilized pellet were both determined, as described (Ganesh et al. 1997). In a separate set of experiments carried out at a final concentration of 5 μM MBP, the sample was centrifuged after variable times of refolding. At each time of folding, the amount of protein in both soluble supernatant and resolubilized pellet were quantitated by UV absorbance at 280 nm and by fluorescence spectroscopy (Ganesh et al. 1997). The results are summarized in Table 1.
Electron microscopy and Congo red dye binding
Amyloid fibrils were prepared by heating 1–2 mM insulin in HCl (pH 2) at 75°C for 4 h. The excess acid was removed, and the precipitate was washed and then resuspended in fresh distilled water. MBP precipitate was prepared in situ by refolding 10 μM protein in 0.1 M GdnHCl (described above) directly on carbon-coated copper grids. Both the samples were negatively stained with 2% (w/v) aqueous uranyl acetate solution and examined in a JOEL 100 CX II electron microscope operated at 80 kV. For the Congo red binding experiments, the insulin precipitates were stained for ∼30 min with freshly prepared 50 μM aqueous Congo red dye solution. MBP precipitation was carried out on a clean glass slide for 20 sec, and then the dye was mixed with the precipitate. The stained samples were covered with a clean glass cover slip and viewed under both normal and polarized light in an optical microscope (magnification 250–400-fold).
Acknowledgments
We are grateful to Dr. M. Hofnung and Prof. Ben de Kruijff for kindly providing the expression plasmids for MBP and SecB, respectively. We thank Vikram Panse for supplying some SecB and advice regarding its purification, A. Guha for providing insulin, and K. Beena for help with the studies on resolubilization of MBP aggregates. We thank Dr. S.S. Indy for electron microscopic studies, and Drs. W.A. Lim and J.S. Weissman for helpful discussions. R.V. and J.B.U. are recipients of Swarnajayanti Fellowships from the Government of India, and of Senior Research Fellowships from the Wellcome Trust. This work was supported by grants from the Departments of Biotechnology, and Science and Technology, Government of India, to R.V. and J.B.U.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
MBP, maltose-binding protein
E. coli, Escherichia coli
GdnHCl, guanidine hydrochloride
CD, circular dichroism
UV, ultraviolet
BSA, bovine serum albumin
RNase A, bovine pancreatic ribonuclease A
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.8101
References
- Agashe, V.R., Shastry, M.C., and Udgaonkar, J.B. 1995. Initial hydrophobic collapse in the folding of barstar. Nature 377 754–757. [DOI] [PubMed] [Google Scholar]
- Anfinsen, C.B. 1973. Principles that govern the folding of protein chains. Science 181 223–230. [DOI] [PubMed] [Google Scholar]
- Betton, J. and Hofnung, M. 1996. Folding of a mutant maltose-binding protein of Escherichia coli which forms inclusion bodies. J. Biol. Chem. 271 8046–8052. [DOI] [PubMed] [Google Scholar]
- Betton, J.M., Sassoon, N., Hofnung, M., and Laurent, M. 1998. Degradation versus aggregation of misfolded maltose-binding protein in the periplasm of Escherichia coli. J. Biol. Chem. 273 8897–8902. [DOI] [PubMed] [Google Scholar]
- Brems, D.N. 1990. Deciphering the second half of the genetic code. In Protein folding (eds. L.M. Gierasch and J. King), pp. 129–135. American Association For The Advancement of Science.
- Breukink, E., Kusters, R., and De Kruijff, B. 1992. In vitro studies on the folding characteristics of the Escherichia coli precursor protein prePhoE. Evidence that SecB prevents the precursor from aggregating by forming a functional complex [published erratum appears in Eur. J. Biochem. 1993 Feb 1;211 (3):909]. Eur. J. Biochem. 208: 419–425. [DOI] [PubMed] [Google Scholar]
- Carrell, R.W. and Lomas, D.A. 1997. Conformational disease. Lancet 350 134–138. [DOI] [PubMed] [Google Scholar]
- Chun, S.Y., Strobel, S., Bassford, Jr., P., and Randall, L.L. 1993. Folding of maltose-binding protein. Evidence for the identity of the rate-determining step in vivo and in vitro. J. Biol. Chem. 268 20855–20862. [PubMed] [Google Scholar]
- Collier, D.N., Bankaitis, V.A., Weiss, J.B., and Bassford, Jr., P.J. 1988. The antifolding activity of SecB promotes the export of the E. coli maltose-binding protein. Cell 53 273–283. [DOI] [PubMed] [Google Scholar]
- di Guan, C., Li, P., Riggs, P.D., and Inouye, H. 1988. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose-binding protein. Gene 67 21–30. [DOI] [PubMed] [Google Scholar]
- Dietzel, I., Kolb, V., and Boos, W. 1978. Pole cap formation in Escherichia coli following induction of the maltose-binding protein. Arch. Microbiol. 118 207–218. [DOI] [PubMed] [Google Scholar]
- Finke, J.M., Gross, L.A., Ho, H.M., Sept, D., Zimm, B.H., and Jennings, P.A. 2000a. Commitment to folded and aggregated states occurs late in interleukin- 1β folding. Biochemistry 39 15633–15642. [DOI] [PubMed] [Google Scholar]
- Finke, J.M., Roy, M., Zimm, B.H., and Jennings, P.A. 2000b. Aggregation events occur prior to stable intermediate formation during refolding of interleukin 1β. Biochemistry 39 575–583. [DOI] [PubMed] [Google Scholar]
- Ganesh, C., Banerjee, A., Shah, A., and Varadarajan, R. 1999. Disordered N-terminal residues affect the folding thermodynamics and kinetics of maltose binding protein. FEBS Lett. 454 307–311. [DOI] [PubMed] [Google Scholar]
- Ganesh, C., Shah, A.N., Swaminathan, C.P., Surolia, A., and Varadarajan, R. 1997. Thermodynamic characterization of the reversible, two-state unfolding of maltose binding protein, a large two-domain protein. Biochemistry 36 5020–5028. [DOI] [PubMed] [Google Scholar]
- Gannon, P.M., Li, P., and Kumamoto, C.A. 1989. The mature portion of Escherichia coli maltose-binding protein (MBP) determines the dependence of MBP on SecB for export. J. Bacteriol. 171 813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, S.J. and Randall, L.L. 1993. Recognition of ligands by SecB, a molecular chaperone involved in bacterial protein export. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 339 343–352; discussion 352–354. [DOI] [PubMed] [Google Scholar]
- Harrison, P.M., Chan, H.S., Prusiner, S.B., and Cohen, F.E. 1999. Thermodynamics of model prions and its implications for the problem of prion protein folding. J. Mol. Biol. 286 593–606. [DOI] [PubMed] [Google Scholar]
- Kapust, R.B. and Waugh, D.S. 1999. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 8 1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar, I., Ravid, M., and Sohar, E. 1976. In vitro synthesis of "amyloid" fibrils from insulin, calcitonin and parathormone. Isr. J. Med. Sci. 12 1137–1140. [PubMed] [Google Scholar]
- Kiefhaber, T., Rudolph, R., Kohler, H.H., and Buchner, J. 1991. Protein aggregation in vitro and in vivo: a quantitative model of the kinetic competition between folding and aggregation. Biotechnology (NY). 9 825–829. [DOI] [PubMed] [Google Scholar]
- King, J., Haase-Pettingell, C., Robinson, A.S., Speed, M., and Mitraki, A. 1996. Thermolabile folding intermediates: Inclusion body precursors and chaperonin substrates. FASEB J. 10 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto, C.A. and Beckwith, J. 1983. Mutations in a new gene, secB, cause defective protein localization in Escherichia coli. J. Bacteriol. 154 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker, S., Lill, R., Ziegelhoffer, T., Georgopoulos, C., Bassford, Jr., P.J., Kumamoto, C.A., and Wickner, W. 1989. Three pure chaperone proteins of Escherichia coli–SecB, trigger factor and GroEL–form soluble complexes with precursor proteins in vitro. EMBO J. 8 2703–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker, S.H., Driessen, A.J., and Wickner, W. 1990. ProOmpA contains secondary and tertiary structure prior to translocation and is shielded from aggregation by association with SecB protein. EMBO J. 9 2309–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiakas, D. and Raina, S. 1997. Protein folding in the bacterial periplasm. J. Bacteriol. 179 2465–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, R.I., Tissieres, A., and Georgepoulos, C. 1994. The biology of heat shock proteins and molecular chaperones. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Panse, V.G., Udgaonkar, J.B., and Varadarajan, R. 1998. SecB binds only to a late native-like intermediate in the folding pathway of barstar and not to the unfolded state. Biochemistry 37 14477–14483. [DOI] [PubMed] [Google Scholar]
- Pecorari, F., Minard, P., Desmadril, M., and Yon, J.M. 1996. Occurrence of transient multimeric species during the refolding of a monomeric protein. J. Biol. Chem. 271 5270–5276. [DOI] [PubMed] [Google Scholar]
- Raffy, S., Sassoon, N., Hofnung, M., and Betton, J.M. 1998. Tertiary structure-dependence of misfolding substitutions in loops of the maltose-binding protein. Protein Sci. 7 2136–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman, B., Ramakrishna, T., and Rao, C.M. 1996. Refolding of denatured and denatured/reduced lysozyme at high concentrations. J. Biol. Chem. 271 17067–17072. [DOI] [PubMed] [Google Scholar]
- Schlunegger, M.P., Bennet, M.J., and Eisenberg, D. 1997. Oligomer formation by 3D domain swapping: A model for protein assembly and misassembly. Adv. Prot. Chem. 50 61–122. [DOI] [PubMed] [Google Scholar]
- Silow, M. and Oliveberg, M. 1997. Transient aggregates in protein folding are easily mistaken for folding intermediates. Proc. Natl. Acad. Sci. 94 6084–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silow, M., Tan, Y.J., Fersht, A.R., and Oliveberg, M. 1999. Formation of short-lived protein aggregates directly from the coil in two-state folding. Biochemistry 38 13006–13012. [DOI] [PubMed] [Google Scholar]
- Sparrer, H., Lilie, H., and Buchner, J. 1996. Dynamics of the GroEL-protein complex: Effects of nucleotides and folding mutants. J. Mol. Biol. 258 74–87. [DOI] [PubMed] [Google Scholar]
- Weiss, J.B., Ray, P.H., and Bassford, Jr., P.J. 1988. Purified secB protein of Escherichia coli retards folding and promotes membrane translocation of the maltose-binding protein in vitro. Proc. Natl. Acad. Sci. 85 8978–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulfing, C. and Pluckthun, A. 1994. Correctly folded T-cell receptor fragments in the periplasm of Escherichia coli. Influence of folding catalysts. J. Mol. Biol. 242 655–669. [DOI] [PubMed] [Google Scholar]
- Xie, Y. and Wetlaufer, D.B. 1996. Control of aggregation in protein refolding: The temperature-leap tactic. Protein Sci. 5 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]