

Abstract
Human angiogenin (Ang) is an unusual homolog of bovine pancreatic RNase A that utilizes its ribonucleolytic activity to induce the formation of new blood vessels. The pyrimidine-binding site of Ang was shown previously to be blocked by glutamine 117, indicating that Ang must undergo a conformational change to bind and cleave RNA. The mechanism and nature of this change are not known, and no Ang-inhibitor complexes have been characterized structurally thus far. Here, we report crystal structures for the complexes of Ang with the inhibitors phosphate and pyrophosphate, and the structure of the complex of the superactive Ang variant Q117G with phosphate, all at 2.0 Å resolution. Phosphate binds to the catalytic site of both Ang and Q117G in essentially the same manner observed in the RNase A–phosphate complex, forming hydrogen bonds with the side chains of His 13, His 114, and Gln 12, and the main chain of Leu 115; it makes an additional interaction with the Lys 40 ammonium group in the Ang complex. One of the phosphate groups of pyrophosphate occupies a similar position. The other phosphate extends toward Gln 117, and lies within hydrogen-bonding distance from the side-chain amide of this residue as well as the imidazole group of His 13 and the main-chain oxygen of Leu 115. The pyrimidine site remains obstructed in all three complex structures, that is, binding to the catalytic center is not sufficient to trigger the conformational change required for catalytic activity, even in the absence of the Gln 117 side chain. The Ang–pyrophosphate complex structure suggests how nucleoside pyrophosphate inhibitors might bind to Ang; this information may be useful for the design of Ang antagonists as potential anti-angiogenic drugs.
Keywords: Ribonuclease A, angiogenin, phosphate binding, X-ray crystallography, inhibitor design
Human angiogenin (Ang) is a 14-kD protein that induces the formation of new blood vessels in vivo (Fett et al. 1985). It is homologous to bovine pancreatic RNase A (Strydom et al. 1985) and exhibits a ribonucleolytic activity that is weak (Shapiro et al. 1986), but nonetheless essential for angiogenic activity (Shapiro and Vallee 1989; Shapiro et al. 1989). Ang was first isolated from culture medium conditioned by adenocarcinoma cells (Fett et al. 1985), and has since been shown to be critical for the establishment and/or metastatic spread of a wide variety of human tumors in athymic mice (Olson et al. 1995; Olson and Fett 1998), most likely by supporting the growth of tumor vasculature. Moreover, clinical studies have revealed increased Ang expression to be associated with progression of several human cancers (Miyake et al. 1999; Shimoyama et al. 1999; Eberle et al. 2000; Etoh et al. 2000; Shimoyama and Kaminishi 2000). These findings identify Ang as a promising target for new anticancer drugs.
The Ang antagonists available at present are proteins (e.g., monoclonal antibodies) or antisense oligonucleotides (Lee and Vallee 1989; Olson et al. 1995; Olson and Fett 1998). Although these agents are effective against tumors in mice and, in some cases, might have therapeutic utility in humans, low molecular weight inhibitors would clearly be advantageous. In light of the central importance of ribonucleolytic activity in Ang, it seems worthwhile to attempt to exploit this feature of the protein in developing such compounds. Toward this end, we have thus far determined a high-resolution crystal structure for free Ang (Acharya et al. 1994; Leonidas et al. 1999a), and identified nucleotide inhibitors [5′-diphosphoadenosine-2′-phosphate (ppA-2′-p) and 2′-deoxyuridine 3′-pyrophosphate (3′→5′) adenosine-2′-phosphate (dUppA-2′-p)] that can potentially serve as starting compounds for structure-based design (Russo et al. 1996a; Russo et al. 2001). However, it has not yet been possible to obtain crystals of Ang in complex with these nucleotides that are suitable for structure determination either by soaking or cocrystallization. The pyrimidine-binding subsite of free Ang is obstructed by the C-terminal segment (see below) and it appears that binding of dUppA-2′-p, and possibly even simpler compounds, would require considerable movement of these residues. In addition, in all crystal forms of Ang grown to date, key phosphate- and purine-binding subsites are occupied by contact residues from neighboring molecules.
In the present study, we have determined the crystal structures of the complexes of Ang with phosphate (Pi) and pyrophosphate (PPi), which are components of the nucleotide inhibitors. Both compounds occupy the catalytic site of Ang without inducing any conformational change. The structure of the complex of phosphate with the superactive Ang variant Q117G (Russo et al. 1994) has also been determined.
Results
The overall structures of the complexes of Ang with Pi and PPi and Q117G–Ang with Pi are similar to that of free Ang [PDB entry 1B1I (Leonidas et al. 1999a)]. In all three structures, the first two residues at the N terminus and last three at the C terminus (121–123) were disordered, as was the side chain of residue 3. There was no side-chain density observed for Gln 19 in the Ang–Pi complex and this residue was therefore modeled as alanine. The root mean square deviations (RMSD) between the positions of the α-carbons in the structures of free Ang and its complexes with Pi and PPi are 0.28 Å and 0.41 Å, respectively; the RMSD for the Q117G–Pi complex versus free Ang is 0.26 Å [as is the RMSD for free Q117G (D.D. Leonidas, R. Shapiro, and K.R. Acharya, unpubl.) versus the Pi complex]. The differences in the structures of the various free proteins and complexes occur primarily in the flexible loop regions and at the N- and C-termini.
The free Ang structure, determined at 1.8 Å resolution (Leonidas et al. 1999a), contains 53 water molecules, whereas the Ang–Pi, Ang–PPi, and Q117G–Pi complexes determined at 2.0 Å resolution contain 46, 104, and 47 water molecules, respectively. A citrate molecule observed in the free Ang structure was not visible in the Ang–PPi complex, although the same crystallization conditions were used; instead, two water molecules were identified at the same position.
In all of the complex structures, the orientations of the residues in the catalytic center in which Pi and PPi bind (i.e., Gln 12, His 13, Lys 40, and His 114) are similar to those in free Ang, although some small differences were observed that appear to optimize interactions with the ligands. In all of the complexes, the electron density maps showed strong positive peaks for the ions (Fig. 1 ▶).
Fig. 1.
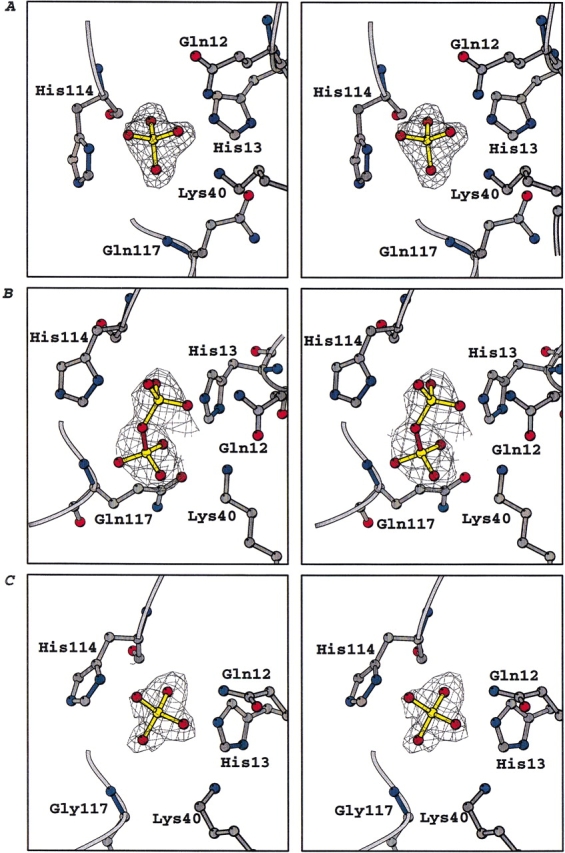
Stereoviews of sigmaA [2|Fo|-|Fc|] electron density maps showing the bound ion in the catalytic site of the Ang–Pi (A), Ang–PPi (B), and Q117G–Pi (C) complexes. The electron density is contoured at 1.0 σ and the figure was created with bobscript (Esnouf 1997).
Interactions of Pi with Ang
The phosphate ion is positioned to hydrogen bond with the side chains of the three catalytic residues His 13, Lys 40, and His 114, as well as with the side chain of Gln 12 and the main-chain N and O of Leu 115 (Fig. 2B ▶, Table 1). This location is essentially the same as that of Pi (Wlodawer et al. 1982) and the phosphate moiety of nucleotide inhibitors observed in crystal structures of RNase A complexes (Leonidas et al. 1997, 1999b; Zegers et al. 1994). Two of the Pi oxygens occupy positions similar to those of water oxygens in free Ang. No water-mediated interactions between Pi and the protein atoms were observed.
Fig. 2.
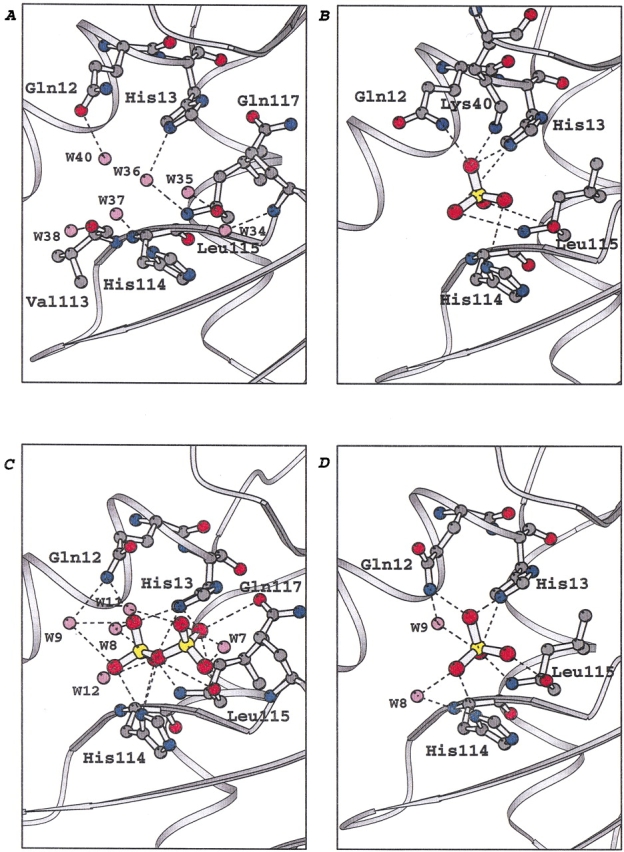
Schematic representations of the potential hydrogen bond interactions with Pi, PPi, and water molecules in the catalytic sites of (A) native Ang, (B) the Ang–Pi complex, (C) the Ang–PPi complex, and (D) the Q117G–Pi complex. Figure was generated with molscript (Kraulis 1991).
Table 1.
Potential hydrogen bond interactions of the ligands in the Ang − Pi, Ang − PPi and Q117G − Pi complexes
| Donor | Acceptor | Ang − Pi | Ang − PPi | Q117G − Pi |
| Gln12 Nɛ2 | PiO4 | 2.64 | — | 2.72 |
| PPiO4 | — | 2.72 | — | |
| His13 Nɛ2 | PiO4 | 3.16 | — | 3.13 |
| PiO2 | 3.19 | — | 2.97 | |
| PPiO7 | — | 3.16 | — | |
| PPiO4 | — | 3.21 | — | |
| PPiO2 | — | 3.06 | — | |
| Lys40 Nζ | PiO4 | 3.21 | — | — |
| His114 Nδ1 | PiO3 | 3.39 | — | 2.79 |
| PPiO2 | — | 3.18 | — | |
| PPiO3 | — | 2.72 | — | |
| PPiO1 | — | 3.23 | — | |
| Leu115 N | PiO2 | 3.03 | — | 2.76 |
| PPiO2 | — | 2.51 | — | |
| PiO1 | Leu115 O | 3.19 | — | 2.91 |
| PPiO2 | — | 2.87 | — | |
| PPiO7 | — | 3.14 | — | |
| Gln117 Oɛ1 | PPiO7 | — | 3.36 | — |
| Water | PPiO1 | — | 2.93 | — |
| Water | PPiO4 | — | 2.34 | — |
| Water | PPiO5 | — | 2.75 | — |
| Water | PPiO3 | — | 3.15 | — |
| Water | PPiO4 | — | 3.21 | — |
| Water | PPiO2 | — | 3.16 | — |
| Water | PPiO4 | — | 3.29 | — |
| Water | PiO3 | — | — | 2.81 |
| Water | PiO2 | — | — | 2.81 |
Numbers in columns are distances in Å. Hydrogen bonds are listed if the distance between a donor and an acceptor is shorter than 3.4 Å and if the angle O-H-N/O is >90°. Hydrogen bond distances were calculated with the program contact (CCP4 version 4.0). Oxygen atoms 1–4 of PPi correspond to the four oxygens of Pi.
Interactions of PPi with Ang
One of the phosphate groups of PPi forms the same set of interactions with the catalytic center of Ang as does Pi, except that the ammonium group of Lys 40 is somewhat beyond hydrogen-bonding distance and there are two additional potential hydrogen bonds with His 114 (Fig. 2C ▶, Table 1). The second phosphate group extends toward Gln 117, making a potential hydrogen bond with the side-chain oxygen of this residue as well as with two atoms (His 13 Nɛ2 and Leu 115 O) that also interact with the first phosphate. The greater number of contacts with Ang formed by PPi as compared with Pi presumably accounts for the sixfold lower Ki of this anion. The inhibitor displaces four of the six water molecules observed in the catalytic site of free Ang (Fig. 2A ▶); the positions of two of the waters are filled by oxygens of the inhibitor. Five water molecules contact PPi, but only one of these also hydrogen bonds to the protein (via Nɛ2 of Gln 12).
Interactions of Pi with Q117G–Ang
The crystal structure of free Ang had revealed a striking and unexpected feature that accounts in part for the low enzymatic activity of this protein: the site that is spatially analogous to the open pyrimidine binding pocket of RNase A is occluded by Gln 117 (Acharya et al. 1994; Leonidas et al. 1999a). Replacement of Gln 117 by Gly or Ala was found to increase the enzymatic activity by 18- to 30-fold (Russo et al. 1994), indicating that this site is also blocked in solution, as was subsequently confirmed by NMR studies (Lequin et al. 1997). The crystal structure of free Q117G–Ang shows no significant changes with respect to the native protein other than the loss of the Gln 117 side chain (D.D. Leonidas, R. Shapiro, and K.R. Acharya, unpubl.). In the present study, the structure of the Q117G–Pi complex was studied to identify possible changes in the interactions at the catalytic site associated with this mutation. The set of potential hydrogen bonds in this complex is identical to those observed in the Ang–Pi complex except that Lys 40 is beyond contact distance (Fig. 2D ▶, Table 1). However, most of the hydrogen bonds are significantly shorter in the Q117G complex. Four of the six water molecules observed in the catalytic site of the free Q117G structure (at 1.8Å resolution, D.D. Leonidas, R. Shapiro, and K.R. Acharya, unpubl.) have been displaced in the complex. The remaining waters mediate interactions of the ligand with the protein (O2 to Gln 12 Nɛ2 and O3 to His 114 Nδ1).
Discussion
Mutational and structural studies on Ang have defined the roles of many active site components. The catalytic residues His 13, Lys 40, and His 114 are positioned almost identically to their counterparts in RNase A, His 12, Lys 41, and His 119, and appear to function in largely the same manner; nonconservative replacements of these amino acids in all cases diminish enzymatic activity by several orders of magnitude (Shapiro and Vallee 1989; Shapiro et al. 1989; Thompson and Raines 1994; Messmore et al. 1995). Therefore, it is not surprising that the two proteins bind Pi quite similarly. However, Ang also contains several unique structural features that suppress its catalytic potency, most notably the obstruction of the putative pyrimidine binding site by side-chain and main-chain atoms of Gln 117 (Acharya et al. 1994; Leonidas et al. 1999a). Modeling results (Russo et al. 1994) indicate that this site must be opened for Ang to bind and cleave RNA. The inactive conformation of the C-terminal segment containing Gln 117 in native Ang is stabilized by several hydrogen bonds (two connecting the side-chain amide of Gln 117 with Thr 44, two between the carboxylate of Asp 116 and Ser 118, and two involving main-chain atoms of the 310 helix 117–121) as well as the hydrophobic burying of Ile 119 and Phe 120. Replacements of these residues individually and in various combinations increase enzymatic activity by up to 30-fold (Harper and Vallee 1988; Russo et al. 1994, 1996b; Shapiro 1998).
The structural rearrangement that opens the pyrimidine binding site of Ang has not yet been defined, and it is not known whether this conformational change occurs prior to substrate binding or is triggered by docking of substrate components outside of the pyrimidine site. Such information should be valuable, and might even be essential, for structure-based design of nucleotide inhibitors of Ang. The present crystal structures show, minimally, that binding of phosphates at the catalytic center of Ang is not sufficient to induce this change, that is, Gln 117 retains its obstructive position in both the Pi and PPi complexes. There is also no reorientation of the C-terminal segment when Pi binds to the Q117G variant. In this regard, it should be noted that the pyrimidine site in Q117G remains obstructed by the main-chain atoms of residue 117, and that a conformational change is still required for RNA cleavage.
The structure of the complex of Ang with PPi may also provide more direct insights that can be used for inhibitor design. The most effective nucleotide inhibitors of Ang identified to date, ppA-2′-p and dUppA-2′-p [Ki = 110 and 150 μM, respectively; (Russo et al. 1996a,b; Russo et al. 2001)], contain pyrophosphate moieties. In the absence of a crystal structure for the complex of Ang with one of these compounds, it would be helpful to know how the pyrophosphate portion might interact with the protein. The crystal structure of the complex of RNaseA with ppA-2′-p [PDB entry 1AFL (Leonidas et al. 1997)] shows that the β-phosphate occupies the catalytic site and the α-phosphate forms two hydrogen bonds with the side chain of Lys 7; this arrangement forces the adenosine to adopt a syn conformation in which the glycosyl torsion angle differs by ∼180 ° from that observed in previous RNase complex structures. However, superposition of the Ang–PPi structure onto that of the RNase A–ppA-2′-p complex (Fig. 3 ▶) reveals that only one of the phosphates (corresponding to the β-phosphate of ppA-2′-p) is bound at an equivalent position. The second phosphate of PPi and the α-phosphate of ppA-2′-p are positioned on opposite sides of this phosphate, toward the pyrimidine site for PPi and toward the purine site for ppA-2′-p. One possible reason for this difference is that the RNase residue that hydrogen bonds with the α-phosphate, Lys 7, is not conserved in Ang. Thus, the α-phosphate of PPi binds on the opposite side because it finds a greater number of hydrogen-bonding interactions with Ang. If the pyrophosphate in the Ang–ppA-2′-p complex binds in the same manner as PPi itself, then the conformation of the adenosine must be anti, rather than syn as in the RNase A complex. The orientation of the adenine ring is an important consideration for the design of modifications to improve binding affinity.
Fig. 3.

Superposition of PPi and ppA-2′-p from the crystal structures of the Ang–PPi (blue), and RNase A–ppA-2′-p (orange) complexes.
Materials and methods
Crystallization
Human Pyr 1–Ang was prepared from a recombinant system in Escherichia coli (Shapiro and Vallee 1992). All crystals were grown at 16°C by use of the vapor diffusion technique. Crystals of Ang in complex with Pi were grown by use of 24% PEG 6000, 0.1 M MES, 0.1 M NaCl (pH 6.0) in the presence of 50 mM disodium hydrogen phosphate. Co-crystals of the Ang–PPi complex were grown using the native conditions (reservoir buffer, 20 mM sodium citrate, 0.2 M sodium potassium tartrate, and 10% PEG 6000 at pH 5.2) (Leonidas et al. 1999a) supplemented with 100 mM sodium pyrophosphate. Orthorhombic crystals of the Q117G variant grown by use of native Ang crystallization conditions were soaked with 200 mM disodium hydrogen phosphate in the reservoir buffer for 24 h. Crystallographic details for the three complexes are presented in Table 2.
Table 2.
Crystallographic statistics
| Dataset | Ang − Pi | Ang − PPi | Q117G − Pi |
| Data collection statistics | |||
| Space group | P21212 | P21212 | P21212 |
| a (Å) | 86.3 | 86.0 | 85.4 |
| b (Å) | 38.5 | 37.8 | 37.5 |
| c (Å) | 33.6 | 33.0 | 38.5 |
| Resolution (Å) | 40–2.0 | 30–2.0 | 40–2.0 |
| Number of reflections measured | 38523 | 53426 | 45669 |
| Number of unique reflections | 7458 | 6973 | 8677 |
| Rsym (%) | 7.6 | 8.5 | 10.1 |
| Completeness (%) | 92.9 | 91.1 | 97.8 |
| (outermost shell) | (97.1) | (78.4) | (85.6) |
| I/σI | 13.2 | 9.6 | 6.4 |
| Refinement statistics | |||
| Rcryst (%) | 18.5 | 23.5 | 19.8 |
| Rfree (%) | 23.0 | 28.7 | 26.0 |
| Number of protein atoms | 992 | 984 | 971 |
| Number of solvent molecules | 46 | 104 | 47 |
| r.m.s. dev. in bond lengths (Å) | 0.009 | 0.009 | 0.010 |
| r.m.s. dev. in bond angles (°) | 1.4 | 1.5 | 1.6 |
| Average B-factor for protein atoms (Å2) | 24.1 | 24.8 | 24.4 |
| Average B-factor for water molecules (Å2) | 35.0 | 42.7 | 34.8 |
| Average B-factor for ligand atoms (Å2) | 52.5 | 47.2 | 48.8 |
Rsym = ∑ (|Ij − 〈I〉|).∑〈I〉 where Ij is the observed intensity of reflection j and 〈I〉 is the average intensity of multiple observations. Rcryst = ∑ ∥Fo | − | Fc ∥ /∑| Fo |, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. 5% of the data which were used for the calculation of Rfree were excluded from the refinement.
Data processing and reduction
Diffraction data for the Ang–Pi and Ang–PPi complexes were collected at station PX 9.5 of the Synchrotron Radiation Source. Data collection for the Pi complex crystal was conducted at room temperature, whereas data for the PPi complex were collected from a flash-frozen crystal by use of the reservoir buffer with 30% glycerol as cryoprotectant. Data for the Q117G–Pi complex were collected at EMBL on beamline X31 at room temperature. All data were processed with denzo and scaled using its companion program scalepack (Otwinowski and Minor 1997; Table 2).
Refinement
The 1.8 Å resolution native Pyr 1–Ang structure (Leonidas et al. 1999a) was used as the starting model for the Ang complexes with Pi and PPi. The coordinates of free Q117G–Ang (D.D. Leonidas, R. Shapiro, and K.R. Acharya, unpubl.) were used for refinement of the Q117G–Pi complex. Alternating cycles of manual model building with the program O (Jones et al. 1991), conventional positional refinement, and simulated annealing refinement as implemented in X-PLOR improved each model, whereas solvent correction as implemented in X-PLOR 3.851 (Jiang and Brünger 1994) allowed all measured data to be used in the refinement. Further refinement of the Ang–PPi complex was performed with the program cns v 0.5 (Brünger et al. 1998). Procedures carried out with CNS included torsion angle dynamics, simulated annealing using a maximum likelihood target function, restrained individual B-factor refinement, conjugate gradient minimization, and bulk solvent correction. The program O was used to adjust the model to fit σA weighted 2 |Fo| - |Fc| electron density maps. During the final stages of refinement, water molecules were inserted into the models at previous positions only if there were peaks in the |Fo| - |Fc| electron density maps with heights greater than 3σ and these were at hydrogen bond forming distances from appropriate atoms. The 2 |Fo| - |Fc| maps were used to verify the presence of these water molecules. Water molecules with a temperature factor >60 Å2 were excluded from subsequent refinement steps. Ligand molecules were included during the final stages of refinement by use of their respective coordinates from the Protein Data Bank. For Pi, the coordinates were obtained from the crystal structure of a phosphate-binding protein in complex with phosphate [PDB entry 1IXH (Wang et al. 1997)] and for PPi the coordinates were obtained from the crystal structure of NH3-dependant NAD synthetase [PDB entry 1NSY (Rizzi et al. 1996)]. The program procheck (CCP4 1994) was used to assess the quality of the final structures. All residues for each of the structures lie in the allowed regions of the Ramachandran (ϕ–ψ) plot. Two peptide bonds connecting residues Ser 37 to Pro 38 and Pro 90 to Pro 91 adopt a cis conformation.
Kinetics
Ki values for inhibition of Ang by Pi and PPi (10 mM and 1.7 mM, respectively) were determined from plots of kcat/Km versus inhibitor concentration as described previously (Shapiro 1998). Assays were performed in 0.2 M MES–NaOH buffer (pH 5.9) at 25°C.
Acknowledgments
This work was supported by the Medical Research Council (Programme grant no. 9540039 to K.R.A.), the Cancer Research Campaign (Project grant no. SP2354/0102 to K.R.A.), the Wellcome Trust (Biomedical Research Collaboration grant no. 044107 to K.R.A. and R.S.), the National Institutes of Health (grant no. HL 52096 to R.S.) and the European Union through its support of the work at EMBL (EU TMR/LSF Grant, Contract Number ERBFMGECT 980134). We thank the staff at the Synchrotron Radiation Sources and EMBL (HASYLAB, c/o DESY, Germany) and members of Acharya laboratory for critical reading of the manuscript.
The atomic coordinates for Ang–Pi, Ang–PPi, and Q117G–Pi complexes have been deposited with the RCSB Protein Data Bank (PDB ID codes are 1HBY, 1H52, and 1H53 respectively).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.13601
References
- Acharya, K.R., Shapiro, R., Allen, S.C., Riordan, J.F., and Vallee, B.L. 1994. Crystal structure of human angiogenin reveals the structural basis for its functional divergence from ribonuclease. Proc. Natl. Acad. Sci. 91 2915–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., GrosseKunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D 50 760–763. [DOI] [PubMed] [Google Scholar]
- Eberle, K., Oberpichler, A., Trantakis, C., Krupp, W., Knupfer. M., Tschesche, H., and Seifert. V. 2000. The expression of angiogenin in tissue samples of different brain tumours and cultured glioma cells. Anticancer Res. 20 1679–1684. [PubMed] [Google Scholar]
- Esnouf R.M. 1997. An extensively modified version of Molscript that includes greatly enhanced coloring capabilities. J. Mol. Graph. 15 132–134. [DOI] [PubMed] [Google Scholar]
- Etoh, T., Shibuta, K., Barnard, G.F., Kitano, S., and Mori, M. 2000. Angiogenin expression in human colorectal cancer: The role of focal macrophage infiltration. Clin. Cancer Res. 6 3545–3551. [PubMed] [Google Scholar]
- Fett, J.W., Strydom, D.J., Lobb, R.R., Alderman, E.M., Bethune, J.L., Riordan, J.F., and Vallee, B.L. 1985. Isolation and characterization of angiogenin, an angiogenic protein from human carcinoma cells. Biochemistry 24 5480–5486. [DOI] [PubMed] [Google Scholar]
- Harper, J.W. and Vallee, B.L. 1988. Mutagenesis of aspartic acid -116 enhances the ribonucleolytic activity and angiogenic potency of angiogenin. Proc. Natl. Acad. Sci. 85 7139–7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, J.-S. and Brünger, A.T. 1994. Protein hydration observed by x-ray diffraction: Solvation properties of penicillopepsin and neuraminidase crystal structures. J. Mol. Biol. 243 100–115. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT - A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lee, F.S. and Vallee, B.L. 1989. Binding of placental ribonuclease inhibitor to the active site of angiogenin. Biochemistry 28 3556–3561. [DOI] [PubMed] [Google Scholar]
- Leonidas, D.D., Shapiro, R., Irons, L.I., Russo, N., and Acharya, K.R. 1997. Crystal structures of ribonuclease A complexes with 5′-diphosphoadenosine 3′-phosphate and 5′-diphosphoadenosine 2′-phosphate at 1.7 Å resolution. Biochemistry 36 5578–5588. [DOI] [PubMed] [Google Scholar]
- Leonidas, D.D., Shapiro, R., Allen, S.C., Subbarao, G.V., Veluraja, K., and Acharya, K.R. 1999a. Refined crystal structures of native human angiogenin and two active site mutants: Implications for the unique functional properties of an enzyme involved in neovascularization during tumour growth. J. Mol. Biol. 285 1209–1233. [DOI] [PubMed] [Google Scholar]
- Leonidas, D.D., Shapiro, R., Irons, L.I., Russo, N., and Acharya, K.R. 1999b. Toward rational design of ribonuclease inhibitors: High resolution crystal structure of a ribonuclease A complex with a potent 3′,5′ pyrophosphate-linked dinucleotide inhibitor. Biochemistry 32 10287–10297. [DOI] [PubMed] [Google Scholar]
- Lequin, O., Thuring, H., Robin, M., and Lallemand, J.-Y. 1997. Three-dimensional solution structure of human angiogenin determined by 1H, 15N-NMR spectroscopy. Characterisation of histidine protonation states and pKa values. Eur. J. Biochem. 250 712–726. [DOI] [PubMed] [Google Scholar]
- Messmore, J.M., Fuchs, D.N., and Raines, R.T. 1995. Ribonuclease A: Revealing structure-function relationships with semisynthesis. J. Am. Chem. Soc. 117 8057–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake, H., Hara, I., Yamanaka, K., Gohji, K., Arakawa, S., and Kamidono, S. 1999. Increased angiogenin expression in the tumor tissue and serum of urothelial carcinoma patients is related to disease progression and recurrence. Cancer 86 316–24. [PubMed] [Google Scholar]
- Olson, K.A. and Fett, J.W. 1998. Inhibition of tumor growth and metastasis by angiogenin antisense therapy. Proc. Amer. Assoc. Cancer Res. 39 98. [Google Scholar]
- Olson, K.A., Fett, J.W., French, T.C., Key, M.E., and Vallee, B.L. 1995. Angiogenin antagonists prevent tumor growth in vivo. Proc. Natl. Acad. Sci. 92 442–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Rizzi, M., Nessi, C., Mattevi, A., Coda, A., Bolognesi, M., and Galizzi, A. 1996. Crystal structure of NH3-dependent NAD+ synthetase from Bacillus subtilis. EMBO J. 15 5125–5134. [PMC free article] [PubMed] [Google Scholar]
- Russo, A., Acharya, K.R., and Shapiro, R. 2001. Small molecule inhibitors of RNase A and related enzymes. Methods Enzymol. (In Press). [DOI] [PubMed]
- Russo, N., Shapiro, R., Acharya, K.R., Riordan, J.F., and Vallee, B.L. 1994. Role of glutamine-117 in the ribonucleolytic activity of human angiogenin. Proc. Natl. Acad. Sci. 91 2920–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, N., Acharya, K.R., Vallee, B.L., and Shapiro, R. 1996a. A combined kinetic and modeling study of the catalytic center subsites of human angiogenin. Proc. Natl. Acad. Sci. 93 804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, N., Nobile, V., DiDonato, A., Riordan, J.F., and Vallee, B.L. 1996b. The C-terminal region of human angiogenin has a dual role in enzymatic activity. Proc. Natl. Acad. Sci. 93 3243–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, R. 1998. Structural features that determine the enzymatic potency and specificity of human angiogenin: Thr-80 and residues 58–70 and 116–123. Biochemistry 37 6847–6856. [DOI] [PubMed] [Google Scholar]
- Shapiro, R. and Vallee, B.L. 1989. Site-directed mutagenesis of histidine-13 and histidine-114 of human angiogenin - Alanine derivatives inhibit angiogenin-induced angiogenesis. Biochemistry 28 7401–7408. [DOI] [PubMed] [Google Scholar]
- ———. 1992. Identification of functional arginines in human angiogenin by site-directed mutagenesis. Biochemistry 31 12477–12485. [DOI] [PubMed] [Google Scholar]
- Shapiro, R., Riordan, J.F., and Vallee, B.L. 1986. Characteristic ribonucleolytic activity of human angiogenin. Biochemistry 25 3527–3532. [DOI] [PubMed] [Google Scholar]
- Shapiro, R., Fox, E.A., and Riordan, J.F. 1989. Role of lysines in human angiogenin - chemical modification and site-directed mutagenesis. Biochemistry 28 1726–1732. [DOI] [PubMed] [Google Scholar]
- Shimoyama, S. and Kaminishi, M.. 2000. Increased angiogenin expression in gastric cancer correlated with cancer progression. J. Cancer Res. Clin. Oncol. 126 468–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoyama, S., Yamasaki, K., Kawahara, M., and Kaminishi, M. 1999. Increased serum angiogenin concentration in colorectal cancer is correlated with cancer progression. Clin. Cancer. Res. 5 1125–1130. [PubMed] [Google Scholar]
- Strydom, D.J., Fett, J.W., Lobb, R.R., Alderman, E.M., Bethune, J.L., Riordan, J.F., and Vallee, B.L. 1985. Amino-acid sequence of human-tumor derived angiogenin. Biochemistry 24 5486–5494. [DOI] [PubMed] [Google Scholar]
- Thompson, J.E. and Raines, R.T. 1994. Value of general acid-base catalysis to ribonuclease A. J. Am. Chem. Soc. 116 5467–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., Luecke, H., Yao, N., and Quiocho, F.A. 1997. A low energy short hydrogen bond in very high resolution structures of protein receptor-phosphate complexes. Nat. Struct. Biol. 4 519–522. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Bott, R., and Sjolin, L. 1982. The refined crystal structure of ribonuclease A at 2.0 Å resolution. J. Biol. Chem. 257 1325–1332. [PubMed] [Google Scholar]
- Zegers, I., Maes, D., Dao-Thi, M.-H., Poortmans, F., Palmer, R., and Wyns, L. 1994. The structures of RNase A complexed with 3′CMP and d(CpA): Active site conformation and conserved water molecules. Protein Sci. 31 2322–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]