

Abstract
Although the crystal structure of Vibrio harveyi luciferase has been elucidated, the binding sites for the flavin mononucleotide and fatty aldehyde substrates are still unknown. The determined location of the phosphate-binding site close to Arg 107 on the α subunit of luciferase is supported here by point mutagenesis. This information, together with previous structure-activity data for the length of the linker connecting the phosphate group to the isoalloxazine ring represent important characteristics of the luciferase-bound conformation of the flavin mononucleotide. A model of the luciferase–flavin complex is developed here using flexible docking supplemented by these structural constraints. The location of the phosphate moiety was used as the anchor in a flexible docking procedure performed by conformation search by using the Monte Carlo minimization approach. The resulting databases of energy-ranked feasible conformations of the luciferase complexes with flavin mononucleotide, ω-phosphopentylflavin, ω-phosphobutylflavin, and ω-phosphopropylflavin were filtered according to the structure-activity profile of these analogs. A unique model was sought not only on energetic criteria but also on the geometric requirement that the isoalloxazine ring of the active flavin analogs must assume a common orientation in the luciferase-binding site, an orientation that is also inaccessible to the inactive flavin analog. The resulting model of the bacterial luciferase–flavin mononucleotide complex is consistent with the experimental data available in the literature. Specifically, the isoalloxazine ring of the flavin mononucleotide interacts with the Ala 74–Ala 75 cis-peptide bond as well as with the Cys 106 side chain in the α subunit of luciferase. The model of the binary complex reveals a distinct cavity suitable for aldehyde binding adjacent to the isoalloxazine ring and flanked by other key residues (His 44 and Trp 250) implicated in the active site.
Keywords: Bacterial luciferase, flavin mononucleotide, flexible docking, structural constraints
Bacterial luciferase, a heterodimeric protein constituted of two homologous subunits (α and β), catalyzes the oxidation of reduced flavin mononucleotide and fatty aldehyde in a process that reduces molecular oxygen to water and releases energy in the form of light (∼490 nm). Both the α (LuxA) and β (LuxB) subunits fold into (β/α)8 barrels in the crystal structures of bacterial luciferase from Vibrio harveyi (Fisher et al. 1995, 1996). The α subunit is primarily responsible for kinetic properties; however, the presence of the β subunit is essential for high catalytic efficiency (Cline and Hastings 1972; Li et al. 1993). The active site thus is believed to be formed by residues in the α subunit with the phosphate moiety of flavin mononucleotide anchored at an electron dense inorganic phosphate site detected on LuxA (Fisher et al. 1995). Another critical factor controlling the bioluminescence activity of bacterial luciferase with reduced flavins is the length of the linker covalently connecting the phosphate moiety and the N10 atom of the isoalloxazine ring (Meighen and MacKenzie 1973). Luciferase activity with flavins containing the isoalloxazine ring connected to the phosphate group by chains of four carbon atoms or longer show full activity whereas a flavin with only three-carbon linker (ω-phosphopropylflavin) has low activity and weak binding affinity similar to riboflavin. Activity with neutral flavins, however, can be stimulated by the addition of inorganic phosphate (Meighen and MacKenzie 1973). Thus, both the location of the phosphate group and its distance from the isoalloxazine ring represent important characteristics of the luciferase-bound conformation of flavin mononucleotide. Here, we combine these experimental constraints with flexible ligand docking to develop a structural model of the flavin mononucleotide complexed to bacterial luciferase.
Results and Discussion
Phosphate-binding site
The electron dense phosphate site on luciferase is within hydrogen-bonding distance to the main chain amide hydrogen atoms of Glu 175 and Ser 176, the hydroxyl group of Thr 179, and the positively charged guanidinium group of Arg 107 on the LuxA subunit (Fisher et al. 1995). An increase in the KM for reduced flavin mononucleotide (FMNH2) as well as a loss in the ability of phosphate to stimulate activity with reduced riboflavin has been reported on replacement of Arg 107 by neutral or negatively charged residues (Moore et al. 1999). We replaced Arg 107 with a positively charged residue (Lys) and found only a small increase (approximately fourfold) in the KM for FMNH2 in comparison to the wild-type luciferase. The relative specificity for FMNH2 over reduced riboflavin remained the same although the activity was decreased (data not shown). More interestingly, the R107K mutation results in a much greater stimulation by phosphate of the bioluminescence activity with reduced riboflavin relative to the wild-type enzyme (Fig. 1 ▶). This increased sensitivity to phosphate for the R107K mutant is consistent with a greater accessibility in the R107K mutant, containing the smaller Lys side chain, to accommodate exogenous phosphate and riboflavin in comparison to the wild-type enzyme, in which the larger Arg side chain normally accommodates the phosphate ester of the riboflavin. Thus, our results provide independent support for the proposed location of the phosphate-binding site. Combined with the previous results (Fisher et al 1995; Moore et al. 1999), it appears highly probable that the 5′ phosphate of FMNH2 (or FMN) is bound to the electrophilic site close to Arg 107 in the LuxA subunit of luciferase.
Fig. 1.

Increase in bioluminescence activity with riboflavin as a function of phosphate concentration with luciferase mutants. Activities were measured using the dithionite assays described in Materials and Methods on injection of decanal into the assay mixture containing the indicated phosphate concentration and 50 μM riboflavin.
Distance between the isoalloxazine ring and the phosphate moiety
The length of the linker connecting the phosphate to the 7,8-dimethyl-isoalloxazine ring is also a critical factor in determining the efficiency of the bioluminescence reaction. Figure 2 ▶ shows that V. harveyi luciferase catalyzes light emission with high efficiency with flavins having four, five, or six, but not three carbon atoms in the linker segment between the phosphate moiety and the isoalloxazine ring (Meighen and MacKenzie 1973). Indeed, the activity of luciferase with reduced ω-phosphopropylflavin is <1% of that with FMNH2 and is similar to that of riboflavin and other neutral flavins lacking the phosphate group (e.g., ω-hydroxylpropylflavin). However, the activity with neutral ω-hydroxylpropylflavin can be stimulated by addition of inorganic phosphate to levels at least 10-fold higher than that with ω-phosphopropylflavin (Meighen and MacKenzie 1973). This result shows that a covalent linker of only three carbon atoms between the isoalloxazine ring and the phosphate group imposes a detrimental structural constraint that may prevent the isoalloxazine ring from adopting the correct geometry in the active site of luciferase. In the separate addition of exogenous inorganic phosphate and ω-hydroxylpropylflavin, the covalent constraint is eliminated thus resulting in higher activity. The unfavorable constraint also is relieved if the covalent linker is increased in length while maintaining its flexibility. The tolerance of the bioluminescence activity to n-alkyl linkers with four to six carbon atoms (Fig. 2 ▶) is likely due to the ability of these flexible linkers to adopt such bioactive conformations that allow a correct binding mode of both the phosphate group and isoalloxazine ring into the luciferase-binding site. Therefore, during the accommodation of the phosphate moiety into the phosphate-binding site and isoalloxazine ring into the adjacent active site, the flavin must have sufficient length in the linker region for the isoalloxazine ring to establish those interactions with luciferase that are productive for the subsequent catalytic steps. These results also showed that the hydroxyl groups in the carbon chain linking the phosphate group to the isoalloxazine ring in flavin mononucleotide are not critical for substrate binding and bioluminescence activity (Meighen and MacKenzie 1973).
Fig. 2.

Relative bioluminescence activities of ω-phosphoalkylflavins with different numbers of carbon atoms linking the phosphate moiety to the isoalloxazine ring. The percentage of the initial light intensity of V. harveyi luciferase with FMN and different fatty aldehydes for each ω-phosphoalkylflavin has been taken from earlier data with the same aldehyde (Meighen and MacKenzie 1973), averaged (plus or minus standard deviation), and then plotted versus the number of carbon atoms in the alkyl chain.
The isoalloxazine ring
The molecular structure of isoalloxazine in the reduced state has been studied since the 1960s and is proposed to vibrate along the axis formed between the N5 and N10 atoms, mimicking the wing movements of a butterfly (Dudley et al. 1964; Tauscher et al. 1973). The atomic structures of several polysubstituted isoalloxazines in the reduced form reveal relatively low bending angles along the N5-N10 axis deviating from planarity by 12.7–35.5 degrees (Norrestam et al. 1969a, 1969b; Leijonmark and Werner 1971; Norrestam and Glehn 1972; Glehn et al. 1977; Porter and Voet 1978). Based on nuclear magnetic resonance (NMR) spectra, Vervoort et al. proposed that the isoalloxazine ring of reduced flavin mononucleotide bound to luciferase is highly planar and rigid (Vervoort et al. 1986). This indicates that the known planar structure of the oxidized isoalloxazine ring would provide an excellent structural model to probe the conformational space of the luciferase-bound reduced flavin mononucleotide.
Modeling the binding mode
A Monte Carlo search of possible binding modes of FMN was conducted as described in Materials and Methods. The large size of the binding cavity allows numerous sterically feasible binding conformations of the flavin ring (not shown). Many of these structures are close in energy, and it is not possible based on the energy values alone to reliably identify a particular conformation as the active binding mode. Figure 3 ▶ shows a plot of root-mean-square deviation (RMSD) of the flavin ring atoms versus relative energy, taking the global minimum-energy structure as the reference for both the RMSD and energy values. We see that the lowest-energy structure is not well separated energetically from other low-energy conformations with significantly different binding modes, that is, with large RMSD values from the global minimum. It is therefore necessary to supplement the energy calculations with additional information to identify the active binding mode.
Fig. 3.

Energetic discrimination of the FMN-binding modes. The root-mean-square deviation of the isoalloxazine ring nonhydrogen atoms (RMSD) is plotted against the relative internal energy (ΔE) of the corresponding luciferase–FMN complexes. The global minimum-energy conformation is taken as reference. (arrows) Two binding modes that are common to the active flavin analogs FMN, ω-phosphobutylflavin, and ω-phosphopentylflavin but inaccessible to the inactive flavin analog ω-phosphopropylflavin. (RMSD) root-mean-square deviation.
For this purpose, we made use of the experimental observations regarding the activity of reduced flavin analogs as a function of the length of the linker between the phosphate and the isoalloxazine ring. As seen in Figure 2 ▶, ω-phosphopropylflavin shows negligible activity whereas ω-phosphobutylflavin and ω-phosphopentylflavin retain full activity (Meighen and MacKenzie 1973). The structural implication is that a three-carbon linker is too short to bridge the phosphate and isoalloxazine-binding sites whereas a four- or five-carbon linker suffices. The comparable activity among FMN and the four- and five-carbon linker flavin analogs further suggests that the isoalloxazine ring is positioned similarly in the bound conformations of these ligands. This provides a strong geometric constraint that can be used to filter the database of conformations obtained in the conformational search of FMN. Specifically, the active conformation of FMN should have the isoalloxazine ring positioned in a manner accessible to FMN, ω-phosphobutylflavin, and ω-phosphopentylflavin but inaccessible to ω-phosphopropylflavin. This geometrical constraint is illustrated schematically in Figure 4 ▶.
Fig. 4.

Geometrical constraints on the bound conformation of the isoalloxazine ring in luciferase. The length of the linker in the flavin analogs restricts the regions of conformational space accessible to the ring. (left) The accessible regions corresponding to each linker length are shown schematically by the three pie-shaped sectors extending from the phosphate anchor. The number labels correspond to the linker length. (right) The active bound conformation of the isoalloxazine ring will be in a region (green) accessible to both the four- and five-carbon linker flavin analogs but inaccessible to the three-carbon linker one.
Conformational search calculations therefore were performed for the bound conformations of ω-phosphopropylflavin, ω-phosphobutylflavin, and ω-phosphopentylflavin by using the same computational protocol used for FMN (see Materials and Methods). Common binding modes among the three ligands then were identified based on the RMSD of the corresponding isoalloxazine ring atoms. At a cutoff of 1.5 Å RMSD, only two binding modes are common to FMN and the four- and five-carbon linker flavin analogs. Comparison of these two structures to those obtained for ω-phosphopropylflavin showed that neither of the two structures was accessible to the isoalloxazine ring of ω-phosphopropylflavin, consistent with the lack of activity of reduced ω-phosphopropylflavin.
The two structures have a relative energy difference of 6.4 kcal/mole with the lower energy one ranked as sixth best in energy among the structures generated in the conformational search on FMN. Examination of the binding mode of the higher energy structure showed relatively weak interactions with the binding site. Its isoalloxazine ring is suspended in the middle of the binding cavity making few close contacts with the surrounding hydrophobic and aromatic residues (Fig. 5 ▶). The expected high mobility of this structure is inconsistent with NMR studies on FMN that indicate that the pyrimidine portion of isoalloxazine is tightly bound and maintains a rigid conformation in the luciferase active site (Vervoort et al. 1986). For these reasons, we discarded the higher energy structure as being unlikely correct. Note also that at a more stringent cutoff of 1.0 Å RMSD, only the lower energy structure remains as common to FMN and the active flavin analogs. The lower energy binding mode common for FMN, ω-phosphobutylflavin, and ω-phosphopentylflavin is shown in Figure 6 ▶.
Fig. 5.

Stereoview of the two binding modes of FMN to luciferase that are common to ω-phosphobutylflavin and ω-phosphopentylflavin, but inaccessible to the inactive ω-phosphopropylflavin. FMN is shown with capped-sticks, with the lower energy conformer colored in white and the higher energy conformer colored in red. The protein atoms that were allowed to move during flexible docking are shown in purple for both complexes. The superposition is based on the remaining part of the protein that was kept rigid during flexible docking (not shown). Hydrogen atoms are omitted for clarity.
Fig. 6.

Stereoview of the lowest energy common binding mode of FMN, ω-phosphobutylflavin, and ω-phosphopentylflavin to the α subunit of bacterial luciferase. The ligand molecules are represented as capped-sticks, with the carbon atoms colored in white for FMN, yellow for ω-phosphobutylflavin, and green for ω-phosphopentylflavin. The protein residues that were allowed to move during flexible docking are shown in purple for all complexes. The alignment is based on the remaining part of the protein that was kept rigid during flexible docking (not shown). Hydrogen atoms are omitted for clarity.
Luciferase–flavin complex
According to the selected lower energy model, the isoalloxazine ring of FMN is predicted to establish several favorable contacts within the luciferase active site (Fig. 7 ▶). The C2 carbonyl oxygen is hydrogen-bonded to the backbone amide NH group of Leu 109 that belongs to a stretch of residues highly conserved among various species homologs of luciferase. The N3 nitrogen atom is in proximity to the thiol group of Cys 106, a residue that has been implicated in affecting the stability of the 4a-hydroperoxylflavin intermediate (Abu-Soud et al. 1993). The C4 carbonyl oxygen is hydrogen-bonded to the backbone amide NH group of Ala 75 that forms a cis-peptide bond with Ala 74. The N5 nitrogen atom is also close (3.7 Å) to the main chain carbonyl oxygen of Ala 74, suggesting that in the reduced form (FMNH2) the N5 hydrogen atom may be hydrogen-bonded to the cis-peptide as well. This interaction could account for the weaker binding of FMN to luciferase compared to FMNH2, although it should be recognized that the addition of the fatty acid product greatly enhances FMN binding (Li and Meighen 1992) leading to dissociation constants for reduced and oxidized flavin that are much more comparable. Nonprolyl cis-peptide linkages occur with very low frequency in proteins (Ramachandran and Mitra 1976; Stewart et al. 1990; Jabs et al. 1999). Thus, the interaction of the Ala 74–Ala 75 cis-peptide bond of luxA with the isoalloxazine ring appears to play an important role in the bioluminescence activity (Fisher et al. 1996). It is also a measure of the success of the modeling strategy presented here that specific interactions between the ligand and this unusual structural feature of the protein were identified. The dimethylbenzene fragment of isoalloxazine makes extensive nonpolar interactions with the side chain of Val 173 and is flanked by the side chains of Phe 6, Ala 74, Ile 191, Ser 193, Trp 194, and Ser 227. Among these residues, mutations of Ser 227 (Chen and Baldwin 1989) and Trp 194 (Li and Meighen 1995) have been shown to affect the kinetics of the bioluminescence reaction. Overall, the model of the luciferase–FMN complex is consistent with the amphipathic character of the isoalloxazine moiety, with the pyrimidine portion interacting with a polar region of the binding site and the dimethylbenzene portion being buried in a hydrophobic protein environment.
Fig. 7.
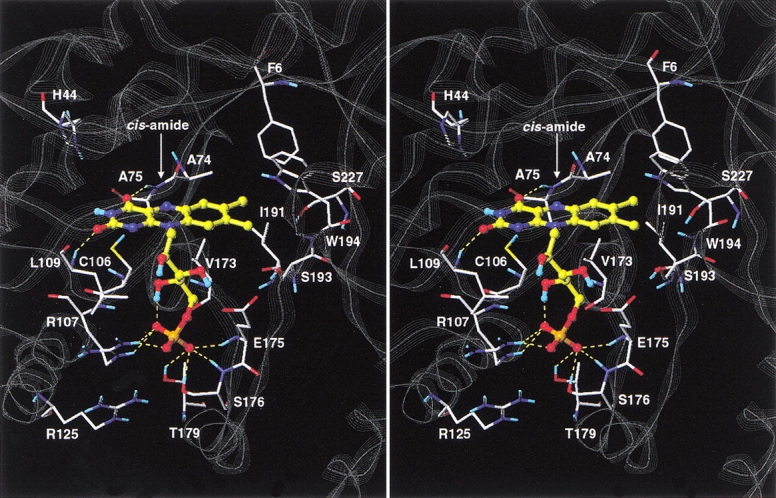
Stereoview of the modeled complex of FMN with the α subunit of bacterial luciferase. The FMN ligand is shown in ball-and-stick representation with the carbon atoms colored in yellow. Selected protein residues lining the binding site and discussed in the text are shown as capped-sticks and labeled. Inter- and intramolecular hydrogen bonds formed by the FMN molecule are shown as yellow dashed lines. Nonpolar hydrogen atoms are omitted for clarity. The backbone of the protein is displayed as a gray line-ribbon.
The phosphate group is engaged in a network of seven intermolecular hydrogen-bonds with the enzyme. Three hydrogen bonds are formed with the guanidinium moiety of Arg 107, two hydrogen bonds with the main chain NH groups of Glu 175 and Ser 176, and two hydrogen bonds with the hydroxyl groups of Ser 176 and Thr 179. The guanidinium moiety of Arg 125 is also in the proximity (reaching as close as 3.8 Å) to the phosphate group. The flexible hydroxylated five-carbon linker of the luciferase-bound FMN deviates from the all-trans geometry with as many as three of the four rotatable C-C bonds being in gauche conformation. The phosphate-proximal hydroxyl group is buried in the complex and forms an intramolecular hydrogen bond with the phosphate moiety. The other two hydroxyl groups of the linker segment are partially exposed to the solvent.
The role of Cys 106 in V. harveyi luciferase has been extensively investigated by both site-directed mutagenesis (Xi et al. 1990; Abu-Soud et al. 1993) and chemical modification studies (Fried and Tu 1984; Paquatte and Tu 1989). Mutation of Cys 106 to valine has been shown to both decrease aldehyde utilization (Xi et al. 1990; Abu-Soud et al. 1993) and destabilize the 4a-hydroperoxylflavin intermediate (Abu-Soud et al. 1993). Because in our model the side chain thiol group of Cys 106 is in close contact with the pyrimidine portion of the isoalloxazine ring of FMN, its mutation to valine would likely alter the polar, complementary protein–ligand interaction occurring at this site. This could result in the destabilization of the 4a-hydroperoxylflavin intermediate and consequently in a decreased ability to oxidize the fatty aldehyde substrate.
His 44 is believed to be implicated as a general base in the catalytic mechanism of bacterial luciferase (Huang and Tu 1997). In our model, the distance between the C4a atom of the isoalloxazine moiety and the Nδ atom of the imidazole ring of His 44 is ∼7 Å, which would accommodate both the hydroperoxyl group of the 4a-hydroperoxylflavin intermediate and the carbonyl group of the aldehyde substrate. Therefore, this structural model of the luciferase–FMN complex suggests that the catalytic reaction would occur on the si-face (Wada et al. 1999) of the isoalloxazine ring facing His 44 side chain. We note that in this region a distinct, spacious cavity is formed between the enzyme and the bound isoalloxazine (Fig. 8 ▶). The many luciferase residues lining this cavity are almost exclusively hydrophobic and include Trp 250 that was proposed to interact with both flavin and aldehyde substrates (Li and Meighen 1995). The present model suggests that only the aldehyde will make direct contact with Trp 250 as it would bind between the isoalloxazine ring of flavin and the indole ring of Trp 250. The large size, the shape, and the hydrophobic nature of this cavity explain why flavin binding is so highly dependent on the presence of a long chain fatty acid, alcohol, or aldehyde (Tu 1979; Li and Meighen 1992).
Fig. 8.

Stereoview of the proposed binding site of the fatty aldehyde substrate onto luciferase–FMN binary complex. The Connolly surface of the luciferase–FMN complex is shown as a mesh and Z-sliced to reveal the cavity protruding inside the protein. Side chains (including Cα atoms) of the luciferase residues lining this cavity are displayed. The arrow indicates the entrance into the cavity. The FMN ligand is shown in ball-and-stick representation with the carbon atoms colored in yellow. A few other key residues discussed in the text also are displayed and labeled for guidance, and hydrogen atoms are omitted for clarity.
Although there is good evidence that the luciferase structure undergoes a conformational change on flavin binding, this has been associated with a protease-sensitive loop of LuxA (AbouKhair et al. 1985). This highly mobile α7a–α7b loop on the surface of the α subunit is largely disordered in the crystal structures of luciferase with and without bound phosphate (Fisher et al. 1995, 1996) and is not present in our model of the luciferase–flavin complex. In addition, its cleavage did not affect the circular dichroism of luciferase (Holtzman et al. 1980), indicating that the structurally well-defined binding site region should remain stable on conformational changes of this loop. Therefore, our flexible docking strategy was built on the assumption of a binding site that is relatively rigid on flavin binding but that responds through limited relaxation to accommodate the conformationally sampled ligand. According to our model of the complex, there appears to be no significant differences before and after flavin binding for most of the residues lining the binding site of luciferase. By superimposing the fixed regions of the FMN-bound structure of luciferase and the energy-minimized structure of apo-luciferase, we calculated an RMSD of 0.87 Å between the positions of equivalent non-hydrogen atoms of 66 binding site residues in the two structures. The largest deviations were found for Glu 175 followed by Leu 109, with RMSDs of 5.70 Å and 2.17 Å, respectively. Exclusion of these two residues resulted in an RMSD of 0.38 Å for the remaining 64 binding site residues. The predicted flavin-induced conformational changes within the binding site of luciferase agree well with the high α subunit temperature factors around residues Leu 109, Met 121, and Glu 175 in the phosphate-free crystal structure, which suggested areas of mobility that could become stationary on flavin binding (Fisher et al. 1996).
In conclusion, we predicted the three-dimensional structure of the bacterial luciferase–flavin mononucleotide complex by using the experimentally determined structural constraints imposed by the anchored phosphate group and the length of the linker connecting the phosphate moiety to the isoalloxazine ring. The combination of the flexible docking with these structural constraints yielded a unique model consistent with other experimental data available in the literature. Elucidation of the chemical mechanism of the bacterial bioluminescence reaction and the role of luciferase in catalysis is perhaps the key goal for understanding light emission in bacteria and the development of this system as a light-emitting sensor. Determination of the relative position of each substrate in the active site of the luciferase is essential to accomplish this goal. Without the crystal structure of the binary and ternary complexes of luciferase with substrates, the proposed model provides one of the only structural frameworks for future investigations on the chemical mechanism.
Materials and methods
Chemicals
Both flavin mononucleotide (FMN) and riboflavin were obtained from Sigma, and the concentrations of flavins were determined using the extinction coefficient of 12,200 M−1 cm−1 at 450 nm. β-Mercaptoethanol was obtained from Sigma. The phosphate buffer solutions (pH 7.0) were prepared by mixing appropriate amounts of NaH2PO4 and K2HPO4 obtained from Baker. Sodium dithionite was purchased from Fisher. Decanal was obtained from Aldrich.
Site-directed mutagenesis
The codon for arginine at position 107 (CGC) was changed to AAA (Lys) and GAA (Glu). The V. harveyi LuxAB gene in M13 was mutated using the Muta-Gene M13 In Vitro Mutagenesis kit from Bio-Rad, and the mutated codons were confirmed by DNA sequencing using the Sequenase DNA Sequencing Kit (version 2) from USB. The mutated LuxAB was transferred to the pT7–5 expression vector, and the sequence was reconfirmed as described above.
Expression and enzyme purification
Each of the pT7–5 plasmids containing the mutated LuxAB genes were transformed into Escherichia coli K38 containing the plasmid pGP1–2 coding for the T7 promoter RNA polymerase expression system (Tabor and Richardson 1985). The cells were grown in the appropriate media containing 100 μg/mL ampicillin and 40 μg/mL kanamycin at 33°C to an OD660 nm of 2.0, centrifuged, and suspended in 10% of the original culture volume of 50 mM phosphate buffer (pH 7.0) and 20 mM β-mercaptoethanol before lysis by ultrasonication. Purification was performed according to previous methods (Gunsalus-Miguel et al. 1972). Protein concentrations were determined using the Bio-Rad protein determination kit with bovine serum albumin as a standard.
Luminescence assays
All luminescence assays were conducted at room temperature (23 ± 2°C) using the dithionite assay (Meighen and Hastings 1971). The activity measurement was conducted at 25°C. The luciferase catalyzed reaction was initiated by injecting 1 mL of 0.1 % decanal (v/v) into 1 mL of assay mixture (pH 7.0) containing luciferase, flavin, 0.2 mg of sodium dithionite, 0.025 M β-mercaptoethanol and phosphate at the indicated concentrations.
Molecular modeling
The 1.5-Å resolution crystal structure of V. harveyi apo-luciferase determined under low-salt conditions (Fisher et al. 1996) and the 2.4-Å resolution crystal structure of the V. harveyi luciferase with an inorganic phosphate ion bound to the α subunit (Fisher et al. 1995) were retrieved from the Protein Data Bank (PDB entries 1LUC and 1BRL, respectively). The apo-luciferase crystal structure (1LUC) was used throughout the molecular modeling experiments, because its improved resolution unravels important structural details not seen in the other crystal structure (e.g., the nonprolyl cis-peptide bond Ala 74–Ala 75). Structure preparation was performed within SYBYL 6.6 molecular modeling software (Tripos, Inc.). Crystallographic water atoms, ethylene glycol molecules, and magnesium ions were removed, and hydrogen atoms were added explicitly. The protonation state at physiological pH was adopted, that is, ionized forms for Arg, Lys, Asp, and Glu residues. Chain termini of the α and β subunits were considered in the ionized state. The anchor termini corresponding to the structurally undefined α7a–α7b loop residues Asp 262 to Arg 290 in the α subunit were capped with acetyl and methylamino groups. A phosphate ion was positioned in the phosphate-binding site on the α subunit as seen in the crystal structure of the luciferase with bound phosphate (1BRL). However, phosphate docking to apo-luciferase was not possible without changing the conformation of the side chain of the Glu 175 residue whose carboxylate group is projected into the phosphate-binding site and overlaps with the phosphate ion. That is, phosphate-binding to luciferase induces a conformational transition in the Glu 175 side chain by displacing the Glu 175 carboxylate from the phosphate-binding site into a solvent exposed location. Therefore, the conformation of the Glu 175 side chain was changed to that observed in the crystal structure of the luciferase with bound phosphate. This luciferase–phosphate complex was structurally refined by energy minimization in SYBYL 6.6 by using the AMBER 4.1 all-atom force field (Cornell et al. 1995) with the Powel minimizer, a distance-dependent (4R) dielectric constant, an 8-Å nonbonded cutoff, and an RMS gradient of 0.05 kcal/mole • Å (these settings also were used throughout the docking experiments). The resulting structure of the luciferase was used for subsequent docking of flavin analogs after removal of the phosphate ion.
Four flavin derivatives were docked individually into the luciferase-binding site: FMN, ω-phosphopentylflavin, ω-phosphobutylflavin, and ω-phosphopropylflavin. A preliminary model first was generated for each flavin analog in complex with luciferase. The FMN molecule was positioned manually with its phosphate group into the phosphate-binding site and the isoalloxazine ring into the adjacent cavity of luciferase (Fisher et al. 1995, 1996) with the connecting linker in all-trans conformation. The complex was energy-minimized with the AMBER 4.1 force field. The FMN atoms as well as the protein residues within 10 Å from FMN were allowed to move during energy minimization. The preliminary complexes with ω-phosphopentylflavin, ω-phosphobutylflavin, and ω-phosphopropylflavin were generated in a similar way. However, the set of mobile protein atoms during energy minimization was kept the same as in the docking of FMN (the largest flavin analog of this series). Missing AMBER 4.1 force field parameters for the isoalloxazine moiety were adapted from those corresponding to parametrized isoalloxazine fragments. This parametrization resulted in a planar geometry of the isoalloxazine ring system. Electrostatic potential-fitted 6–31 G* partial atomic charges of the ligands were calculated in Gaussian 94 (Gaussian, Inc.) on the AMBER 4.1 optimized all-trans geometries. The preliminary complexes were used to initialize the flexible docking of flavin analogs to luciferase.
Flexible docking was performed by conformational search using a Monte Carlo with energy-minimization procedure (Li and Scheraga 1987; Nägler et al. 1999; Therrien et al. 2001). In each Monte Carlo minimization cycle, a starting conformation of the protein–ligand complex was generated by randomly perturbing one or more dihedral angles of the ligand molecule while keeping its phosphate group as an anchor. This starting conformation then was subjected to an AMBER 4.1 energy minimization in which the ligand molecule and a constant, predefined set of protein residues (also used to generate the preliminary luciferase–FMN complex; see above) were allowed to move. The decision of accepting or rejecting the resulting conformation of the complex was taken on the energy basis according to the Metropolis probability criterion. An accepted conformation or a rejected conformation whose internal energy was within 10 kcal/mole above the internal energy of current accepted conformation was stored in a database after passing through the chirality and RMSD-based conformer redundancy filters. All rotatable bonds of a ligand were subjected to random perturbations for a total of 1000 Monte Carlo minimization cycles. The resulting databases of energy-ranked feasible conformations of the luciferase complexes were submitted to pharmacophoric mapping by using the activity data of the flavin analogs.
Acknowledgments
We thank Dr. Robert E. MacKenzie (McGill University, Canada) for informative and helpful discussions concerning the structures of flavin analogs used in this work. This work is supported by Grant MT4314 from the Medical Research Council of Canada and a Max Stern fellowship (to L.Y.-C.L.). This is National Research Council of Canada publication number 44786.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
FMN, flavin mononucleotide
FMNH2, reduced FMN
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.7201
References
- AbouKhair, N.K., Ziegler, M.M., and Baldwin, T.O. 1985. Bacterial luciferase: Demonstration of a catalytically competent altered conformational state following a single turnover. Biochemistry 24 3942–3947. [DOI] [PubMed] [Google Scholar]
- Abu-Soud, H.M., Clark, A.C., Francisco, W.A., Baldwin, T.O., and Raushel, F.M. 1993. Kinetic destabilization of the hydroperoxy flavin intermediate by site-directed modification of the reactive thiol in bacterial luciferase. J. Biol. Chem. 268 7699–7706. [PubMed] [Google Scholar]
- Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, Jr. K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W., and Kollman, P.A. 1995. A second generation force field for the simulation of protein, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117 5179–5197. [Google Scholar]
- Chen, L.H. and Baldwin, T.O. 1989. Random and site-directed mutagenesis of bacterial luciferase: Investigation of the aldehyde binding site. Biochemistry 28 2684–2689. [DOI] [PubMed] [Google Scholar]
- Cline, T.W. and Hastings, J.W. 1972. Mutationally altered bacterial luciferase. Implications for subunit functions. Biochemistry 11 3359–3370. [DOI] [PubMed] [Google Scholar]
- Dudley, K.H., Ehrenberg, A., Hemmerich, P., and Müller, F. 1964. Spektren und Strukturen der am Flavin-Redoxsystem beteiligten Partikeln. Helv. Chim. Acta 47 1354–1383. [Google Scholar]
- Fisher, A.J., Raushel, F.M., Baldwin, T.O., and Rayment, I. 1995. Three-dimensional structure of bacterial luciferase from Vibrio harveyi at 2.4 Å resolution. Biochemistry 34 6581–6586. [DOI] [PubMed] [Google Scholar]
- Fisher, A.J., Thompson, T.B., Thoden, J.B., Baldwin, T.O., and Rayment, I. 1996. The 1.5-Å resolution crystal structure of bacterial luciferase in low salt conditions. J. Biol. Chem. 271 21956–21968. [DOI] [PubMed] [Google Scholar]
- Fried, A. and Tu, S.C. 1984. Affinity labeling of the aldehyde site of bacterial luciferase. J. Biol. Chem. 259 10754–10759. [PubMed] [Google Scholar]
- Glehn, M.V., Stensland, B., and Gartner, B. 1977. 5–(N-benzyl-N-methyliminioprop-2-enyl)-3,7,8,10-tetramethyl-1,5-dihydroisoalloxazine perchlorate, a model compound of a flavoprotein inhibitor complex. Acta Cryst. B33 2388–2391. [Google Scholar]
- Gunsalus-Miguel, A., Meighen, E.A., Nicoli, M.Z., Nealson, K.H., and Hastings, J.W. 1972. Purification and properties of bacterial luciferases. J. Biol. Chem. 247 398–404. [PubMed] [Google Scholar]
- Holzman, T.F., Riley, P.L., and Baldwin, T.O. 1980. Inactivation of luciferase from the luminous marine bacterium Beneckea harveyi by proteases: Evidence for a protease labile region and properties of the protein following inactivation. Arch Biochem. Biophys. 205 554–563. [DOI] [PubMed] [Google Scholar]
- Huang, S. and Tu, S.C. 1997. Identification and characterization of a catalytic base in bacterial luciferase by chemical rescue of a dark mutant. Biochemistry 36 14609–14615. [DOI] [PubMed] [Google Scholar]
- Jabs, A., Weiss, M.S., and Hilgenfeld, R. 1999. Non-proline cis peptide bonds in proteins. J. Mol. Biol. 286 291–304. [DOI] [PubMed] [Google Scholar]
- Leijonmarck, M. and Werner, P. 1971. Studies on flavin derivatives. The crystal structure of 5-acetyl-9-bromo-1,3,7,8-tetramethyl-1,5-dihydroalloxazine. Acta Chem. Scand. 25 2273–2290. [Google Scholar]
- Li, Z. and Meighen, E.A. 1992. Fatty acid-enhanced binding of flavin mononucleotide to bacterial luciferase measured by steady-state fluorescence. Biochem. Biophys. Res. Commun. 188 497–502. [DOI] [PubMed] [Google Scholar]
- ———. 1995. Tryptophan 250 on the α subunit plays an important role in flavin and aldehyde binding to bacterial luciferase. Effects of W → Y mutations on catalytic function. Biochemistry 34 15084–15090. [DOI] [PubMed] [Google Scholar]
- Li, Z. and Scheraga, H.A. 1987. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. 84 6611–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., Szittner, R., and Meighen, E.A. 1993. Subunit interactions and the role of the luxA polypeptide in controlling thermal stability and catalytic properties in recombinant luciferase hybrids. Biochim. Biophys. Acta 1158 137–145. [DOI] [PubMed] [Google Scholar]
- Meighen, E.A. and Hastings, J.W. 1971. Binding site determination from kinetic data. Reduced flavin mononucleotide binding to bacterial luciferase. J. Biol. Chem. 246 7666–7674. [PubMed] [Google Scholar]
- Meighen, E.A. and MacKenzie, R.E. 1973. Flavine specificity of enzyme-substrate intermediates in the bacterial bioluminescent reaction. Structural requirements of the flavine side chain. Biochemistry 12 1482–1491. [DOI] [PubMed] [Google Scholar]
- Moore, C., Lei, B., and Tu, S.C. 1999. Relationship between the conserved α subunit arginine 107 and effects of phosphate on the activity and stability of Vibrio harveyi luciferase. Arch Biochem. Biophys. 370 45–50. [DOI] [PubMed] [Google Scholar]
- Nägler, D.K., Zhang, R., Tam, W., Sulea, T., Purisima, E.O., and Ménard, R. 1999. Human cathepsin X: A cysteine protease with unique carboxypeptidase activity. Biochemistry 38 12648–12654. [DOI] [PubMed] [Google Scholar]
- Norrestam, R. and Glehn, M.V. 1972. Studies on flavin derivatives. The crystal and molecular structure of 9-bromo-1,3,7,8,10-pentamethyl-1,5-dihydroisoalloxazine. Acta Cryst. B28 434–440. [Google Scholar]
- Norrestam, R., Glehn, M.V., Hagman, L.O., and Kierkegaard, P. 1969a. Studies on flavin derivatives. The crystal and molecular structure of 9-bromo-5-hydro-1,3,7,8,10-pentamethyl-1,5-dihydroalloxazine. Acta Chem. Scand. 23 2199–2201. [Google Scholar]
- Norrestam, R., Kierkegaard, P., Stensland, B., and Torbjornsson, L. 1969b. 5-Acetyl-3,7,8,10-tetramethyl-1,5-dihydroalloxazine: Crystal structure and extended Hückel calculations for different molecular geometries. Chem. Commun. 1227 1250–1251. [Google Scholar]
- Paquatte, O. and Tu, S.C. 1989. Chemical modification and characterization of the α cysteine 106 at the Vibrio harveyi luciferase active center. Photochem. Photobiol. 50 817–825. [DOI] [PubMed] [Google Scholar]
- Porter, D.J.T. and Voet, D. 1978. The crystal structure of a flavin-nicotinamide biscoenzyme in two oxidation states. Models for flavin-nicotinamide interactions. Acta. Cryst. B34 598–610. [Google Scholar]
- Ramachandran, G.N. and Mitra, A.K. 1976. An explanation for the rare occurrence of cis peptide units in proteins and polypeptides. J. Mol. Biol. 107 85–92. [DOI] [PubMed] [Google Scholar]
- Stewart, D.E., Sarkar, A., and Wampler, J.E. 1990. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 214 253–260. [DOI] [PubMed] [Google Scholar]
- Tabor, S. and Richardson, C.C. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. 82 1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauscher, L., Ghisla, S., and Hemmerich, P. 1973. NMR-study of nitrogen inversion and conformation of 1,5-dihydro-isoalloxazines ("reduced flavin"). Helv. Chim. Acta 56 630–644. [DOI] [PubMed] [Google Scholar]
- Therrien, C., Lachance, P., Sulea, T., Purisima, E.O., Qi, H., Ziomek, E., Alvarez-Hernandez, A., Roush, W.R., and Ménard, R. 2001. Cathepsins X and B can be differentiated through their respective mono- and dipeptidyl carboxypeptidase activities. Biochemistry 40 2702–2711. [DOI] [PubMed] [Google Scholar]
- Tu, S.C. 1979. Isolation and properties of bacterial luciferase-oxygenated flavin intermediate complexed with long-chain alcohols. Biochemistry 18 5940–5945. [DOI] [PubMed] [Google Scholar]
- Vervoort, J., Muller, F., O'Kane, D.J., Lee, J., and Bacher, A. 1986. Bacterial luciferase: A carbon-13, nitrogen-15, and phosphorus-31 nuclear magnetic resonance investigation. Biochemistry 25 8067–8075. [DOI] [PubMed] [Google Scholar]
- Wada, N., Sugimoto, T., Watanabe, H., and Tu, S.C. 1999. Computational analysis of the oxygen addition at the C4a site of reduced flavin in the bacterial luciferase bioluminescence reaction. Photochem. Photobiol. 70 116–122. [PubMed] [Google Scholar]
- Xi, L., Cho, K.W., Herndon, M.E., and Tu, S.C. 1990. Elicitation of an oxidase activity in bacterial luciferase by site-directed mutation of a noncatalytic residue. J. Biol. Chem. 265 4200–4203. [PubMed] [Google Scholar]