

Abstract
The platelet integrin αIIbβ3 is representative of a class of heterodimeric receptors that upon activation bind extracellular macromolecular ligands and form signaling clusters. This study examined how occupancy of αIIbβ3's fibrinogen binding site affected the receptor's solution structure and stability. Eptifibatide, an integrin antagonist developed to treat cardiovascular disease, served as a high-affinity, monovalent model ligand with fibrinogen-like selectivity for αIIbβ3. Eptifibatide binding promptly and reversibly perturbed the conformation of the αIIbβ3 complex. Ligand-specific decreases in its diffusion and sedimentation coefficient were observed at near-stoichiometric eptifibatide concentrations, in contrast to the receptor-perturbing effects of RGD ligands that we previously observed only at a 70-fold molar excess. Eptifibatide promoted αIIbβ3 dimerization 10-fold more effectively than less selective RGD ligands, as determined by sedimentation equilibrium. Eptifibatide-bound integrin receptors displayed an ectodomain separation and enhanced assembly of dimers and larger oligomers linked through their stalk regions, as seen by transmission electron microscopy. Ligation with eptifibatide protected αIIbβ3 from SDS-induced subunit dissociation, an effect on electrophoretic mobility not seen with RGD ligands. Despite its distinct cleft, the open conformer resisted guanidine unfolding as effectively as the ligand-free integrin. Thus, we provide the first demonstration that binding a monovalent ligand to αIIbβ3's extracellular fibrinogen-recognition site stabilizes the receptor's open conformation and enhances self-association through its distant transmembrane and/or cytoplasmic domains. By showing how eptifibatide and RGD peptides, ligands with distinct binding sites, each affects αIIbβ3's conformation, our findings provide new mechanistic insights into ligand-linked integrin activation, clustering and signaling.
Keywords: Integrins, fibrinogen receptor, light scattering, analytical ultracentrifugation, electron microscopy, molecular modeling, ligand binding, hydrodynamics
The αIIbβ3 integrin is the prototypical member of a family of integral membrane proteins that maintain communication and contact between a cell's interior and its external environment (Critchley et al. 1999; Giancotti and Ruoslahti 1999; Hynes 1992; Plow et al. 2000). Like many integrins, the 226-kD αIIbβ3 complex must be activated before it can function as a high-affinity receptor for adhesive proteins (Hughes and Pfaff 1998; Shattil et al. 1998; Liddington and Bankston 2000;). Ordinarily, αIIbβ3 resides embedded in the plasma membrane of human blood platelets where potentially saturating concentrations of its primary physiological ligand, the 340-kD protein fibrinogen, surround its globular ligand-recognition domain (Phillips et al. 1988; Shattil 1999). However, contacts are only established following vascular injury, when internal molecular messengers initiate conformational changes (Sims et al. 1991) that are transmitted outward from its cytoplasmic regions, through its entwined membrane-spanning polypeptides (Du et al. 1993), and to its large extracellular domain. These processes lead to a functional integrin capable of fibrinogen binding (Ginsberg et al. 1992).
Like other integrins, αIIbβ3 can signal bidirectionally (Dedhar and Hannigan 1996). Ligand binding to its ectodomain soon links the 15-nm-distant cytoplasmic domain and the actin cytoskeleton (Hughes and Pfaff 1998; Giancotti and Ruoslahti 1999). Conformational changes in the αIIbβ3 receptor, including increased self-association or clustering, are considered to be involved in this process of outside-in signaling (Shattil et al. 1998; Giancotti and Ruoslahti, 1999; Hantgan et al. 1999). Although high-resolution structural data are still emerging (Leahy 1997; Dickeson and Santoro 1998; Emsley et al. 2000), much remains to be learned about the molecular bases for integrin activation, ligand binding, and signal transduction.
αIIbβ3's extracellular domain contains distinct yet interacting ligand-binding sites with specificity for fibronectin or fibrinogen (Wippler et al. 1994; Peterson et al. 1998; Hu et al. 1999). Synthetic peptides that contain fibronectin's Arg-Gly-Asp-Ser (RGDS) integrin-targeting motif (Ruoslahti and Pierschbacher 1987) can be cross-linked to a narrow segment of the β3 chain (D'Souza et al. 1988). In contrast, peptides homologous to the critical HHLGGAKQAGDV integrin-recognition site found at the carboxyl termini of fibrinogen's γ chains (Kloczewiak et al. 1989; Farrell et al. 1992; Hettasch et al. 1992; Weisel et al. 1992) bind primarily to a region on the αIIb subunit (Santoro and Lawing Jr. 1987; D'Souza et al. 1990). The observation, by surface plasmon resonance, that prebound fibrinogen can be readily displaced from αIIbβ3 by an RGD ligand provides direct evidence that fibrinogen's binding site is separate from, but allosterically linked to, its RGD site (Hu et al. 1999). This conclusion has recently been supported by the demonstration, using human/rat αIIbβ3 chimeras, that sequences within αIIb's third and fourth amino-terminal repeats regulate the ability of RGDS to block fibrinogen binding (Basani et al. 2001).
It has been proposed that steric hindrance between a ligand-recognition region (termed the A-domain [Lee et al. 1995a,b) or β-I-like domain (Leitinger and Hogg 2000)] on the β3 subunit and a putative β-propeller fold on the αIIb subunit (Huang and Springer 1997; Springer 1997) prevents these macromolecular ligands from binding to either site in the receptor's inactive state (Loftus and Liddington 1997). However, low molecular weight RGD peptides can bind to the β3 subunit on the resting integrin (D'Souza et al. 1988) and block access by their parent adhesive proteins (Plow et al. 1985). We have recently shown that RGDX ligands (X = Ser, Trp, Phe) shift a conformational equilibrium toward an open integrin, thus providing a mechanistic explanation for their effects on αIIbβ3's conformation and activity (Hantgan et al. 1999). The present study extends those observations by examining the structural consequences of ligation of αIIbβ3's fibrinogen binding site. Because the binding sites for RGD ligands and fibrinogen are spatially distinct yet coupled allosterically (Hu et al. 1999; Basani et al. 2001) we hypothesized that binding a fibrinogen-mimetic would have a significant impact on αIIbβ3's tertiary and quaternary structure.
Functional and biophysical studies have been reported with the fibrinogen-derived peptide HHLGGAKQAGDV (Parise et al. 1987; Hawiger et al. 1989; Hawiger 1995; Erb et al. 1997). However, the millimolar concentrations required to affect αIIbβ3 conformation (Parise et al. 1987; Erb et al. 1997) suggest this flexible synthetic peptide (Donahue et al. 1994; Ware et al. 1999) may not accurately model the integrin recognition site on the γ-domain of the native fibrinogen molecule (Weisel et al. 1992). In this study, eptifibatide (see below) served as a high-affinity ligand mimetic with fibrinogen-like selectivity for αIIbβ3 (Phillips and Scarborough 1997). We have followed an integrated biophysical, ultrastructural, and molecular modeling approach to investigate the relationship between occupancy of αIIbβ3's fibrinogen binding site and the receptor's solution structure and stability.
Eptifibatide (N6-(aminoiminomethyl)-N2-(3-mercapto-1oxopropyl - L - lysylglycyl - L - α - aspartyl - L - tryptophanyl L-prolyl-cysteinamide, cyclic (1–6)-disulfide) is the generic name for Integrilin (COR Therapeutics), a recently approved pharmaceutical integrin antagonist used to treat cardiovascular disease by blocking platelet:fibrinogen adhesive interactions (Phillips and Scarborough 1997; Goa and Noble 1999). Eptifibatide's design was initially based on the Lys-Gly-Asp (KGD) site on barbourin, a 73-residue snake venom peptide that binds tightly to the αIIbβ3 integrin but not to αvβ3 (Scarborough et al. 1991). Eptifibatide was designed to retain similar selectivity for αIIbβ3 by incorporating a lysine derivative, homoarginine, into its structure. A recent molecular mechanics study indicates that a key tryptophan present at equivalent positions in both barbourin and eptifibatide also contributes to their fibrinogen-like selectivity for αIIbβ3 (Minoux et al. 2000). The net result is a conformationally constrained heptapeptide that at low micromolar concentrations blocks fibrinogen binding to the αIIbβ3 receptor but not the αvβ3 integrin (Scarborough et al. 1993a,b; Phillips and Scarborough 1997; Goa and Noble 1999).
We will show that near-stoichiometric eptifibatide concentrations trigger a multistep process in αIIbβ3 that starts with a conformational change in its extracellular domain and leads to integrin self-association, mediated at least in part through its distant transmembrane and/or cytoplasmic regions. We will also show that eptifibatide binding stabilizes noncovalent interactions between the αIIb and β3 subunits, an effect not observed with RGDX ligands. By showing how both eptifibatide and RGDX peptides perturb αIIbβ3's structure, our findings provide a new structural framework for understanding the dynamic processes of integrin affinity regulation and ligand-induced signal transduction, as well as new insights into the mechanism of a class of cardiovascular-disease drugs known as integrin antagonists.
Results
Effects of eptifibatide on αIIbβ3 solution conformation
Molecular models of eptifibatide and the control cyclic peptide used in this study are shown in Figure 1 ▶. Despite the overall chemical and structural similarity of these conformationally constrained peptides, they differed markedly in their biological activity and, as will be shown in the sections that follow, in their effects on αIIbβ3 structure. The bioactivity of each peptide (dissolved in buffered saline) was measured in a platelet aggregation assay and the data analyzed by nonlinear regression to determine the peptide concentration required to reduce the initial rate of aggregation to 50% of its control value, denoted the IC50 (Hantgan et al. 1992). Eptifibatide exhibited an IC50 = 0.24 ± 0.06 μM, whereas control peptide had no significant effect on platelet aggregation at concentrations up to 100 μM.
Fig. 1.

Molecular models of eptifibatide and control cyclic peptide. (A) Eptifibatide, (N6-(aminoiminomethyl)-N2-(3-mercapto-1-oxopropyl-L-lysylglycyl-L-α-aspartyl-L-tryptophanyl-L-prolyl-cysteinamide, cyclic (1–6)-disulfide). (B) Inactive control peptide, cyclo-L-cysteinyl-L-lysyl-D-alanyl-L-aspartyl-L-tryptophanyl-L-prolyl-L-cystinyl-amide (CKADWPC). Both models were prepared and minimum-energy configurations obtained with ALCHEMY and SYBYL molecular graphics/analysis software (Tripos, Inc.).
Light-scattering measurements
The effects of eptifibatide on αIIbβ3's biophysical parameters were examined to determine how fibrinogen receptor occupancy effected the integrin's conformation. Dynamic and static light-scattering data collected from dilute solutions of αIIbβ3 (2.7–3.8 μM) yielded D20,w = 2.74 ± 0.05 F and Mw = 226 ± 29 K, respectively, consistent with the receptor's asymmetric structure (Hantgan et al. 1993, 1999). Addition of eptifibatide (3 μM) to achieve a 10% molar excess over αIIbβ3 decreased D20,w by ∼10% in <15 min, with no significant increase in the weight-average molecular weight. As shown in Figure 2 ▶ (solid triangles), similar decreases in D20,w (0.904 ± 0.009 times control, P < 0.001, n = 4) were obtained at eptifibatide concentrations ranging from 3–140 μM. In contrast, addition of control cyclic peptide (open triangles) caused little or no change (0.978 ± 0.022 times control, n = 2). Each data point is an average of 6–8 values obtained within 90 min. of peptide addition. The solid line was calculated from hydrodynamic theory for multisubunit particles (De La Torre and Bloomfield 1981; Spotorno et al. 1997), utilizing bead models of the closed and open forms of the integrin, which predict a 7% change in the frictional coefficient upon ligation (Hantgan et al. 1999), coupled with our ligand-linked isomerization and oligomerization model. An eptifibatide binding constant of 0.2 μM and an integrin association constant of 3 × 104 M−1, which predicts ∼5% dimer formation, were employed in this simulation.
Fig. 2.

Fractional change in αIIbβ3 integrin's translational diffusion and sedimentation coefficients as a function of ligand concentration. Diffusion coefficients were determined by dynamic light-scattering measurements of the (90 °) intensity autocorrelation function of αIIbβ3 (in the presence and absence of ligand). Data were analyzed by the method of cumulants, following correction for solvent contributions. Error bars denote the standard deviation of replicate measurements (n = 6–8) performed with each sample. Solid triangles, eptifibatide; open triangles, control cyclic peptide. Sedimentation velocity data were analyzed with SVEDBERG to obtain weight-average sedimentation coefficients as a function of ligand concentration. Solid circles, eptifibatide; open circles, control cyclic peptide. Because the errors were typically <0.005 S, the error bars fall within the symbols. The solid and dashed lines were obtained from simulations of the changes in D20,w calculated for bead models utilizing hydrodynamic theory for multisubunit particles (De La Torre and Bloomfield 1981; Spotorno et al. 1997) and a ligand-linked isomerization and oligomerization model (Hantgan et al. 1999). In both cases, the ligand dissociation constant KL = 0.2 μM. The solid line was obtained using an occupied receptor self-association constant Ka = 0.03 L/μM, whereas Ka = 0 for the dashed line (to simulate isomerization without oligomerization).
Sedimentation velocity measurements
The effects of eptifibatide on αIIbβ3's solution structure were also examined by sedimentation velocity analyses. Sedimentation velocity determinations were performed with the αIIbβ3 integrin alone, in the presence of eptifibatide and control cyclic peptide. Analyses with SVEDBERG software yielded a weight-average sedimentation coefficient, s20, w = 8.35 ± 0.15 S (n = 5) for αIIbβ3 alone (1.6–4.0 μM) and a similar value, 8.31 ± 0.04 S (n = 2), with control cyclic peptide (10 and 100 μM). In contrast, significantly smaller sedimentation coefficients were obtained in the presence of eptifibatide (10 and 100 μM), namely, 7.88 ± 0.12 S (n = 6, P = 0.001 vs. integrin alone). As shown in Figure 2 ▶, analysis of the complete dataset indicated that eptifibatide (10 and 100 μM, open symbols) reduced αIIbβ3's sedimentation coefficient to 0.947 ± 0.017 times the control value. In contrast, this ratio was 0.986 ± 0.022 (n = 2) with control cyclic peptide (10 and 100 μM; open symbols). The dashed line, which closely follows the experimental data obtained with eptifibatide, was again calculated from hydrodynamic theory for multisubunit particles (De La Torre and Bloomfield 1981; Spotorno et al. 1997) coupled with our ligand-linked isomerization model (Hantgan et al. 1999). The difference in magnitude between the ligand-induced changes observed by sedimentation velocity and dynamic light scattering are most likely due to the increased sensitivity of the latter technique to small quantities of oligomers (Hantgan et al. 1999).
Evidence for reversible conformational changes
Because sedimentation velocity provided a more precise measure of eptifibatide-induced conformational changes, this technique was used to test for reversibility. Figure 3 ▶ depicts analyses of sedimentation velocity profiles obtained with DCDT+ software; results are presented as g(s*) profiles (i.e., weight-average distributions of sedimenting species [Stafford III 1992]). Data were collected with αIIbβ3 alone (open symbols and dashed line), in the presence of 10 μM eptifibatide (solid symbols and solid line) and with a sample incubated with 10 μM eptifibatide for 60 min and then separated free of ligand by rapid size exclusion chromatography (Hantgan et al. 1999; dark gray symbols and dashed-dot line). Note the shift in the peak of the g(s*) profile from 8.71 S to 8.20 S induced by eptifibatide. This effect was substantially reversed following ligand removal, as these data can now be described by a single species at 8.51 S. Averaging results obtained in this and a replicate transient ligand exposure experiment indicated 79 ± 9% of eptifibatide's effects on αIIbβ3's sedimentation coefficient were reversed following ligand removal.
Fig. 3.

Effects of transient exposure to ligand on the distribution of sedimenting species observed with αIIbβ3. Sedimentation velocity data for αIIbβ3 alone (open circles and dashed line) and in the presence of eptifibatide (10 μM, solid circles and solid line) were analyzed with DCDT+ time derivative software (J. Philo) to obtain sedimentation coefficient distribution functions, g(s*) vs. s* (Stafford III 1992). Solid lines were obtained by fitting the resultant distribution functions to a single ideal species. In addition, a separate αIIbβ3 sample (dark gray circles and dashed-dot line) was isolated free of eptifibatide following a 60-min incubation at 23°C prior to the onset of the sedimentation velocity run. Note how the shift observed in the presence of eptifibatide was substantially reversed following ligand removal.
Effects of eptifibatide on αIIbβ3 self-association
Sedimentation equilibrium measurements with αIIbβ3 alone (3.2–3.8 μM) and in the presence of eptifibatide (3–106 μM) tested the hypothesis that fibrinogen receptor occupancy promotes integrin self-association. The data were analyzed initially in terms of a single ideal species model to obtain a set of weight-average molecular weight parameters. In the absence of ligands, αIIbβ3 exhibited Mw = 221 ± 5 K (Fig. 4 ▶), a value in excellent agreement with our previous results (Hantgan et al. 1999). Addition of a stoichiometric concentration of eptifibatide caused a sharp increase in Mw to 301 ± 8 K; similar values were obtained over the range 3–106 μM eptifibatide (Fig. 4 ▶, solid symbols). In contrast, adding control cyclic peptide (10 and 100 μM) yielded no significant changes in Mw (Fig. 4 ▶, open symbols)
Fig. 4.

Effects of eptifibatide on αIIbβ3 molecular weight distribution determined by sedimentation equilibrium measurements. Sedimentation equilibrium data obtained with αIIbβ3 as a function of the concentration of eptifibatide (solid symbols) or control cyclic peptide (open symbols) were analyzed with WinNONLIN (Johnson et al. 1981) to obtain the weight-average molecular weight of the free and ligand-bound receptor. Additional analyses were performed with data from the αIIbβ3:eptifibatide samples to obtain the receptor's self-association constant, Ka = 5.9 × 104 L/mole. The solid line was obtained with an isomerization and oligomerization model (Hantgan et al. 1999), as described in the Fig. 2 ▶ legend, with KL = 0.2 μM and this value of Ka.
Next, the complete dataset obtained with eptifibatide was analyzed globally to obtain an estimate of the association constant for dimerization of the liganded receptor, yielding Ka = 5.9 ± 0.9 × 104 M−1. Computation of the predicted change in Mw using this value of Ka in a ligand-linked isomerization and oligomerization model (Hantgan et al. 1999) yielded the solid line shown in Figure 4 ▶, which closely follows the experimentally determined molecular weight values. This simulation corresponds to an equilibrium mixture of αIIbβ3 at 3 μM containing some 22% dimers and 5% trimers. We note that this oligomer fraction exceeds those observed either by dynamic light scattering or sedimentation velocity (Fig. 2 ▶), suggesting that additional αIIbβ3 self-association took place in the 48 h required to complete the sedimentation equilibrium experiments. However, because comparable weight-average molecular weights were obtained at two rotor speeds, the oligomerization process appears reversible (Johnson et al. 1981).
Electron microscopy analyses of the effects of eptifibatide on αIIbβ3 structure
The preceding biophysical analyses indicated that eptifibatide binding to αIIbβ3 causes a change to a more open conformation and increases integrin self-association. These conclusions are supported by electron microscopy examination of integrin:eptifibatide complexes. Complexes of αIIbβ3 were diluted in 0.05 M ammonium formate buffer at pH 7.4, 30 mM octyl glucoside, 30% glycerol to a final concentration of 20–25 μg/ml, sprayed onto freshly cleaved mica and shadowed with tungsten. Glycerol is necessary to prevent drying artifacts, but its concentration was lowered considerably from previous experiments to minimize its effects on αIIbβ3 oligomerization (Hantgan et al. 1999). The resultant αIIbβ3 preparations displayed mostly single particles (>85%) with a globular head about 8 × 12 nm and two tails about 15 nm long projecting from one side and usually joined distally (Fig. 5A ▶), features we and others have observed previously for unligated integrins (Carrell et al. 1985; Weisel et al. 1992; Hantgan et al. 1999).
Fig. 5.

A Gallery of electron microscope images of rotary shadowed αIIbβ3 complexes, alone and in the presence of eptifibatide or control cyclic peptide. (A) Examples of αIIbβ3 images obtained in the absence of peptides. Individual αIIbβ3 complexes are characterized by a globular head with two long tails that are most often joined at their tips. (B) Examples of αIIbβ3 images obtained in the presence of control cyclic peptide (10 uM). Here the molecular features of the αIIbβ3 complexes closely resemble those seen without peptide (as in panel A). (C) Examples of αIIbβ3 complexes obtained in the presence of eptifibatide (10 uM). In most cases, a separation of the globular head into two distinct nodules can be seen. (D) Examples of αIIbβ3 dimers observed in the presence of eptifibatide (10 uM). Dimers joined tail to tail were the most common structure seen, as detailed in the text. Bar = 50 nm. Interpretation of these electron micrographs may be aided by schematics of the closed, open, and oligomeric integrins in Fig. 8 ▶.
Eptifibatide at a final concentration of 10 μM was incubated with αIIbβ3 at a concentration of 350 μg/mL for 0.5 h prior to shadowing. Examination of the complexes by electron microscopy after incubation with eptifibatide revealed that there was often a separation of the heads in many complexes (Fig. 5C ▶). In other words, two smaller, separate nodules were observed instead of a large, single nodule. On average, about 55% of integrin complexes appeared to be made up of two separated nodules (n = 400).
In interpreting these results, some cautionary notes are in order. These molecular features are near the resolution of this technique, so that very good preparations were necessary to observe the separation, and it could have been missed in some cases. Also, the separation was more difficult to detect in oligomers of αIIbβ3, which were common in these preparations, as noted below. Finally, the separation might not be visible in complexes that lie on the mica surface in certain orientations. For all of these reasons, the estimate of molecules showing a conformational change is an underestimate. As a control, 10 μM of a cyclic control peptide was used (Fig. 5B ▶). Here, the images were essentially the same as those from experiments without any additions (Fig. 5A ▶). Some complexes, however, appeared to have separated heads in all preparations. With no peptide, about 10% had separated heads; whereas with cyclic control peptide, 9% did.
An increase in self-association of αIIbβ3 particles was also observed in the presence of eptifibatide. Because the αIIbβ3 complexes aggregate in the absence of detergent, 30 mM octyl glucoside was used in all experiments, including during dilution prior to spraying. Nevertheless, some oligomers, generally dimers with tail-to-tail interactions, were always present. With no peptide, 14% of complexes were present as dimers. The amount of self-association increased strikingly in the presence of eptifibatide but not in the presence of the control peptide, in which only 15% were seen as dimers. In the presence of eptifibatide, the percentage of total αIIbβ3 particles present in aggregates was 65%. Of these, 87% were dimers, 8% were trimers and 5% were present as larger particles. Dimers were oriented 180° apart with the distal ends of their tails (transmembrane or cytoplasmic domains) interacting (Fig. 5D ▶); whereas larger oligomers were arranged as rosettes (Weisel et al. 1992). Approximately 48% (n = 400) of the oligomers exhibited separated nodules in their head regions, although this is most likely an underestimate, as noted earlier. In addition, the visualization of the open form is somewhat more difficult in the aggregates, so the determination of the percentage of integrins in the open conformation is more accurate from the individual particles.
Interpretation of the images in Figure 5 ▶ may be aided by our integrin models, based both on electron microscopy and hydrodynamic data, that will be presented in the Discussion.
Effects of eptifibatide on αIIbβ3 stability
Subunit association
Because the ligated form of αIIbβ3 exhibited a subunit separation visible by electron microscopy, we were surprised to find this new conformer was resistant to SDS-induced subunit dissociation. This observation was made in the course of routine electrophoretic analyses of αIIbβ3 samples obtained at the beginning and end of the preceding biophysical characterizations. As shown in Figure 6A ▶, lanes 1 and 2, control integrin samples incubated in 1% SDS at room temperature showed two prominent bands at molecular masses of 134 ± 2 kD and 100 ± 2 kD, corresponding to the αIIb and β3 subunits, respectively. In contrast, samples incubated for as little as 2 h with 3 μM eptifibatide showed a major new band at an apparent molecular mass of 180 ± 2 kD (Fig. 6A ▶, lanes 3, 4). The ∼180-kD species was consistently observed in αIIbβ3 samples incubated with eptifibatide (3–140 μM/2–80 h) and accounted for 67 ± 18% (n = 22) of the average staining intensity of the αIIb and β3 subunits. The ∼180-kD band was not observed in αIIbβ3 samples incubated with control cyclic peptide (e.g., Fig. 6A ▶, lanes 5, 6). Western blotting demonstrated that the ∼180-kD band observed with eptifibatide-containing samples exhibited immunoreactivity with antibodies specific for both the αIIb and β3 subunits (data not shown).
Fig. 6.

Electrophoretic analyses of the effects of eptifibatide on αIIbβ3 subunit structure. (A) Samples of αIIbβ3 obtained before/after sedimentation velocity measurements (as described in Fig. 3 ▶'s legend) were denatured with sodium dodecyl sulfate, subjected to polyacrylamide gel electrophoresis, and then stained with Coomassie Brilliant Blue to visualize the polypeptides. Lanes 1, 2: αIIbβ3; 3, 4: +10 μM eptifibatide; 5, 6: +10 μM control cyclic peptide. Note the characteristic αIIb & β3 doublet (molecular masses ∼134 kD and ∼100 kD) in lanes 1, 2 and 5,6, as well as the additional ∼180-kD band seen with the eptifibatide sample. Molecular-mass markers are shown to the left of the gel photograph. (B) Samples of αIIbβ3 obtained in the transient-exposure-to-ligand experiment (described in Fig. 3 ▶'s legend) were subjected to electrophoretic analyses, as described above. Note the appearance of the ∼180-kD species in αIIbβ3 samples containing eptifibatide (3, 4) and the decrease in this band's intensity ∼6 h after ligand removal (compare lanes 5 and 6). (C) Temperature-dependent changes in the relative staining intensity of the ∼180-kD band observed with integrin + eptifibatide samples. Samples were incubated at the indicated temperature for 1 h in the presence of 1% SDS (but no reducing agent) prior to electrophoretic analysis and Coomassie Brilliant Blue staining. (Inset) Integrin + eptifibatide samples incubated in 1% SDS at 23°C (lane 1) and 40°C (lane 2) prior to electrophoretic analysis.
Like the conformational change detected by sedimentation velocity, the effects of eptifibatide on αIIbβ3's electrophoretic mobility were reversible following ligand removal. For example, Figure 6B ▶ shows analyses of samples obtained during the course of the sedimentation velocity experiments depicted in Figure 3 ▶. The ∼180-kD species was not seen with αIIbβ3 alone (Fig. 6B ▶, lanes 1, 2) but accounted for 66 ± 3% of the staining intensity in an αIIbβ3 sample incubated for 1–8 h with 10 μM eptifibatide (Fig. 6B ▶, lanes 3, 4). Whereas a similar level (65%) was observed 30 min after ligand removal (lane 5), by 6 h, the band intensity had decreased to 24% (lane 6). In a replicate experiment, the ∼180-kD band decreased from 78% to 31% over a 7-h period.
The ∼180-kD band was absent following disulfide bond reduction (data not shown), raising the possibility that it could represent a transient cystine-linked integrin conformer. However, we also determined that the intensity of the ∼180-kD band decreased markedly following incubation of integrin + eptifibatide samples in 1% SDS at elevated temperatures in the absence of reducing agent. For example, Fig. 6C ▶ (insert, lane 1] shows a strongly staining ∼180-kD band in an αIIbβ3:eptifibatide sample that was incubated in SDS for 1h at 23°C. In contrast, the band is not seen in a similar sample incubated for 1h at 40°C (lane 2). As shown in Fig. 6C ▶, the intensity of the ∼180-kD species decreased with increasing temperature over the range 23°C–40°C, exhibiting a melting point of ∼30°C. Additional studies (data not shown) showed that preincubation of integrin samples with excess fluorescein maleimide prior to addition of eptifibatide had no effect on the intensity of the ∼180-kD band. These observations argue against a thiol exchange mechanism and indicate that ligation with eptifibatide provides partial protection against SDS-induced subunit dissociation through noncovalent stabilization of the αIIbβ3 complex.
Global stability
The influence of eptifibatide on αIIbβ3 conformation and stability were examined in more detail by a series of UV-absorbance and intrinsic fluorescence measurements. αIIbβ3 exhibited a broad absorbance band with a maximum extinction coefficient at 277.5 nm (Fig. 7A ▶, solid line), both alone and in the presence of 10 μM eptifibatide. Fluorescence intensity measurements (λex 278 nm) of αIIbβ3 alone exhibited an emission maximum (λem) at 341 nm (Fig. 7B ▶, solid line). These observations indicate that in the absence of ligands, αIIbβ3 exhibited a fluorescence emission spectrum dominated by its 24 tryptophan residues, although 55 tyrosines are also present (Fitzgerald et al. 1987; Poncz et al. 1987;). Addition of 10 μM eptifibatide caused an ∼19% decrease in fluorescence intensity with λem now at 340 nm (not shown). However, a similar decrease in intensity (∼24%) was observed in the presence of 10 μM control cyclic peptide, suggesting that an inner filter effect due to ∼20% increased absorbance at the excitation wavelength in the presence of these tryptophan-containing peptides was responsible for the decreased fluorescence (Cantor and Schimmel 1980).
Fig. 7.

Unfolding of the αIIβ3 complex monitored by changes in intrinsic fluorescence intensity. (A) Because these samples exhibited an isosbestic wavelength at 277.5 nm, an excitation wavelength of 278 nm (1-nm bandwidth) was used for fluorescence measurements and the emission signal normalized by the absorbance of each sample at 277.5 nm to correct for small differences in protein concentration. (B) Fluorescence emission spectra obtained with αIIβ3 alone (solid line) and in the presence of 4 M guanidinium chloride (dashed line). Note how guanidinium chloride decreased the fluorescence signal by ∼31% and shifted the emission maximum to 344 nm. (C) Guanidinium chloride denaturation profiles obtained with αIIbβ3 alone (solid symbols) and in the presence of eptifibatide (open symbols). The fraction unfolded was determined from the changes in normalized fluorescence intensity measured as a function of denaturant concentration. The combined dataset was fit to a cooperative unfolding model to obtain the solid line, characterized by a transition midpoint at ∼1.8 M guandinium chloride.
In contrast, addition of 4 M guanidinium chloride (GdnCl) shifted αIIbβ3's absorbance maximum to 276 nm (Fig. 7A ▶, dashed line); the resultant difference spectrum was maximum at 286 nm (∼5% increased extinction coefficient), and an isosbestic point at 277.5 nm was observed. Under these denaturing conditions, αIIbβ3 exhibited a red-shifted fluorescence emission maximum at 344 nm and a 31% decrease in intensity (Fig. 7B ▶, dashed line).
These observations formed the basis for monitoring unfolding of the αIIbβ3 complex in the presence/absence of eptifibatide. In particular, fluorescence emission data were normalized by the absorbance at 278 nm to provide an index (F*) of the concentration-corrected fluorescence emission spectra. These data were analyzed as follows to obtain U, the fraction unfolded, as a function of the [GdnCl]:
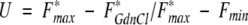 |
1 |
where the subscripts max and min refer to the normalized fluorescence emission obtained at 0 and 4 M GdnCl, respectively, whereas the term F*GdnCl defines the emission at a particular denaturant concentration.
Fig. 7C ▶ shows GdnCl denaturation profiles obtained with αIIbβ3 (0.6–1.4 μM) alone (solid symbols) and in the presence of excess eptifibatide (open symbols, 3.4 & 10.1 μM). The solid line was obtained by fitting the combined data (± eptifibatide) to a cooperative unfolding model with a midpoint [GdnCl]mid at 1.8 ± 0.18 M GdnCl (where b is an empirical index of cooperativity):
 |
2 |
Fitting each dataset separately yielded unfolding midpoints at 1.69 ± 0.35 and 1.85 ± 0.22 M GdnCl in the presence/absence of eptifibatide, respectively. These observations indicate that eptifibatide binding does not destabilize the αIIbβ3 complex, despite the major rearrangements it induces in integrin conformation.
Discussion
This work is the first to show that occupancy of the fibrinogen-binding site on the αIIbβ3 integrin with a monovalent ligand initiates a multistep process involving structural changes in the receptor's extracellular domain. These changes are transmitted over distances up to 15 nm to enhance integrin clustering through its transmembrane and/or cytoplasmic domains. We have shown by laser light scattering, analytical ultracentrifugation, and electron microscopy that near-stoichiometric concentrations of eptifibatide, a cyclized heptapeptide with fibrinogen-like selectivity for αIIbβ3, shift a conformational equilibrium toward an open integrin. Despite its distinct cleft, its cooperative guanidinium chloride-induced unfolding profile shows this eptifibatide-bound conformer retains the structural stability of the ligand-free integrin. Electrophoretic analyses showed that eptifibatide binding actually protects the αIIbβ3 complex from SDS-induced subunit dissociation, an effect not seen with RGDX peptides (Hantgan et al. 1999). Both sedimentation equilibrium measurements and electron microscopy observations show that ligation with eptifibatide enhances the formation of integrin clusters more effectively than β3-targeted RGD ligands (Hantgan et al. 1999).
Figure 8 ▶ presents a ligand-linked conformational change and oligomerization scheme (solid arrows) that, when coupled with hydrodynamic modeling, quantitatively describes the results we have obtained with eptifibatide, as well as those we previously reported with RGDX peptides (Hantgan et al. 1999). By showing that eptifibatide and RGD ligands induce similar conformational changes in αIIbβ3's ectodomain, our findings reinforce the concept that the receptor's fibrinogen binding site and its RGD site are allosterically linked (Hu et al. 1999; Basani et al. 2001). However, as will be explored in detail below, these ligands exhibited important differences in their effects on αIIb/β3 subunit interactions and integrin oligomerization.
Fig. 8.

Ligand-linked integrin conformational change and oligomerization model. Illustration of proposed scheme with beads models of the closed and open αIIbβ3 integrin conformations, as well as integrin dimers (Hantgan et al. 1999). Note the similarity of these models to the electron micrographs in Fig. 6 ▶. The αIIb heavy chain is represented by purple beads while the αIIb light chain is shown as blue beads; the β3 subunit is depicted as a string of yellow beads. The white sphere represents an octyl glucoside micelle. The β3 chain region to which RGD peptides have been cross-linked (D'Souza et al. 1988) is represented by a red bead; the αIIb region where KQAGDV peptides have been cross-linked (D'Souza et al. 1990) is represented by a pink bead. Computation of the changes in hydrodynamic parameters (Hantgan et al. 1999) for the scheme indicated by the solid arrows formed the basis of the simulations in Figs. 2 and 4 ▶ ▶ and helped to understand the shifts in sedimentation velocity in Fig. 3 ▶. However, we recognize the possibility that intermediates interacting through their ectodomains, such as those indicated in the dotted boxes, may also form but may collapse to more elongated dimeric structures through micelle fusion and/or due to surface interactions during preparation for electron microscopy. This scheme describes the effects of both eptifibatide (this work) and RGD ligands (Hantgan et al. 1999) on αIIbβ3 conformation, thus underscoring the allosteric linkage between their distant binding sites (Hu et al. 1999; Basani et al. 2001).
Like its RGDX-induced counterparts, the new conformer we have observed in the presence of eptifibatide exhibits the hallmark of an activated integrin, namely, a distinct subunit separation in the receptor's extracellular domain (Loftus and Liddington 1997; Hantgan et al. 1999; Emsley et al. 2000). The conformational change alone exposes a cavity large enough to accommodate fibrinogen's γ domain (Yee et al. 1997), the subunit that harbors the key KQAGDV integrin-targeting sequence on αIIbβ3's primary physiological ligand (Kloczewiak et al. 1989; Farrell et al. 1992; Hettasch et al. 1992). In addition to these conformational changes, we have found that eptifibatide promotes the formation of integrin dimers and trimers. In fact, the self-association constant of ∼6 × 104 M−1 obtained from sedimentation equilibrium data indicates eptifibatide was at least 10-fold more effective at increasing αIIbβ3 oligomerization than any of the RGDX ligands we had previously characterized (Hantgan et al. 1999). The αIIbβ3 clustering induced by eptifibatide is especially interesting in light of the report that fibrinogen both activated and caused clustering of αIIbβ3 in lipid bilayers; whereas the monovalent ligands GRGDS and the fibrinogen γ-chain dodecapeptide caused activation only (Erb et al. 1997).
Eptifibatide's ability to change αIIbβ3's conformation and actively promote its self-association may explain the interplay between ligand binding to its ectodomain, receptor clustering and the regulation of signal transduction (Miyamoto et al. 1995; Erb et al. 1997; Giancotti and Ruoslahti 1999; Humphries 1999). Precedent for long-range propagation of conformational changes comes from studies of signaling mechanisms in the aspartate receptor, a member of the two-transmembrane-helix receptor family (Koshland 1998; Ottemann et al. 1999). These investigators recently proposed a piston model that explains how aspartate binding to the receptor's periplasmic domain induces a conformational change in a transmembrane helix that is transmitted to the ∼10 nm distant cytoplasmic domain (Ottemann et al. 1999). However, other transmembrane signaling modes they proposed, including the scissors and seesaw models may be more appropriate for the αIIbβ3 integrin, given the conformational changes observed here by electron microscopy. The scissors model is especially attractive because it provides a mechanical linkage that can explain bidirectional integrin signaling (O'Toole et al. 1994). We propose that by binding to αIIbβ3's ectodomain, ligand-mimetics such as eptifibatide (this study) and RGDX peptides (Hantgan et al. 1999) perturb the interactions between subunits, by analogy, opening the scissors by pulling on the blades on the receptor's extracellular face, separating its distant transmembrane and/or cytoplasmic regions and making them available for oligomerization. Thus the conformational change they induce in αIIbβ3's ectodomain may be similar to that induced physiologically by events starting from the cytoplasmic domains (inside-out signaling). Likewise, the receptor clustering that results from eptifibatide's occupancy of the integrin's fibrinogen-binding site may mimic outside-in signaling.
The integrin dimers observed in electron micrographs appear to be joined at their tails, suggesting that transmembrane and/or cytoplasmic domain interactions may play a significant role in oligomerization. These concepts are supported by NMR data (Vinogradova et al. 2000) and molecular models (Haas and Plow 1997) that have identified multiple interaction sites on the carboxy-terminal regions of the αIIb and β3 subunits. Furthermore, dimerization of transmembrane helices in detergent micelles has been carefully documented for glycophorin A using both NMR and small angle x-ray scattering (MacKenzie et al. 1997; Bu and Engelman 1999). However, it also is possible that the integrin dimers we observed were initially stabilized by ectodomain interactions lost following octyl glucoside micelle fusion (VanAken et al. 1986; Lorber et al. 1990) and/or surface interactions during preparation for electron microscopy. Thus, we present hypothetical side-by-side dimeric integrin intermediates within the dotted boxes in Figure 8 ▶. Precedent for ligand-induced oligomers joined through their extracellular domains comes from studies with hematopoietic receptor complexes, such as the multimers that form when the cytokine IL-6 binds to its receptor's α and β subunits (Wells and deVos 1996).
Eptifibatide is a newly approved cardiovascular-disease drug, an integrin antagonist, and our observations may explain key aspects of its pharmaceutical activity (Phillips and Scarborough 1997). We propose that eptifibatide perturbs αIIbβ3's conformation and blocks receptor occupancy by its primary physiological ligand, fibrinogen, thus preventing platelet aggregation (Phillips et al. 1988). Eptifibatide's effects are specific, as no significant changes in αIIbβ3's hydrodynamic or electrophoretic parameters were obtained with the biologically inactive control peptide. As illustrated in Figure 1A ▶, eptifibatide displays a cup-shaped configuration similar to that described for other high-affinity integrin antagonists (Minoux et al. 1998, 2000). Interestingly, as shown in Figure 1B ▶, this feature, which may be important for receptor recognition, is interrupted by a kink in the biologically inactive control cyclic peptide.
Because eptifibatide also shares key structural elements with a peptide analog of the fibrinogen γ chain site necessary for integrin recognition (Hawiger 1995; Suehiro and Plow 1997), our results have special significance for understanding the relationship between receptor occupancy and integrin conformation. In particular, cyclo (S,S) KYGCHarDWPC behaves as a high-affinity analog of the KQADGV site on fibrinogen's γ chains (Ware et al. 1999); whereas cyclo (S,S) KYGCRGDWPC mimics fibrinogen's RGD sequences (Suehiro and Plow 1997; Cierniewski et al. 1999). These cyclized ligands bind to distinct regions of the αIIbβ3 receptor separated by ∼6–9 nm (Cierniewski et al. 1999). Our ligated integrin model (Fig. 8 ▶) indicates that the RGD cross-linking site on the β3 chain (D'Souza et al. 1988) is now separated by ∼9 nm from the γ chain binding domain on the αIIb subunit (D'Souza et al. 1990). In the absence of ligands, our model predicts only a 2-nm subunit separation (Hantgan et al. 1999).
Despite this cleft, we have found that the open conformer is not a denatured species. In particular, we have shown that upon eptifibatide binding, αIIbβ3 shows increased stability toward SDS-induced subunit separation and a cooperative guandinium chloride unfolding profile similar to the native integrin. Eptifibatide's ability to stabilize αIIbβ3 subunit interactions against SDS-induced dissociation seems similar to the recently reported effects of echistatin on other RGD-dependent integrins (Thibault 2000). However, our findings extend Thibault's observations by showing that mild heating disrupts the SDS-stable integrin:eptifibatide complexes. Our data also argue against a disulfide-exchange activation mechanism (Yan and Smith 2001), as eptifibatide-induced complexes were observed even in the presence of excess thiol-blocking reagent. Additional evidence for a noncovalent process comes from our finding that eptifibatide's stabilizing effects on αIIbβ3 are readily overcome by guanidinium chloride, which denatures proteins without disrupting their disulfide bonds (Creighton 1984).
In conclusion, we suggest that the geometric constraints imposed by insertion of αIIbβ3's stalks into the lipid bilayer may promote clusters of ligand-activated integrins stabilized by additional interactions of their ectodomains, such as those in Figure 8 ▶. Such integrin multimers have recently been observed in a 21 Å-resolution structure of the αvβ5:adenovirus complex obtained by cryoelectron microscopy (Chiu et al. 1999). While high-resolution data are still emerging, our integrated biophysical, electron microscope, and molecular modeling approach has furthered development of a structural scaffold that can explain the conformational differences between resting and activated integrins, as well the mechanisms of outside-in signaling (Clark and Brugge 1995; Loftus and Liddington 1997; Hughes and Pfaff 1998; Giancotti and Ruoslahti 1999; Leisner et al. 1999).
Materials and methods
Purification and characterization of the αIIbβ3 complex
The platelet integrin receptor αIIbβ3 was isolated from the detergent-solubilized membrane fraction of human blood platelets (American Red Cross) by lentil lectin affinity chromatography and size exclusion chromatography, as previously described in detail (Hantgan et al. 1993). This procedure yields milligram quantities of nearly monodisperse αIIbβ3 in a pH 7.4 buffer (HSC-OG) containing 0.13 M NaCl, 0.01 M HEPES, 0.002 M CaCl2, 3 × 10–8 M basic trypsin inhibitor (Aprotinin, Sigma), 10−6 M leupeptin, and 0.03 M n-octyl-β-D-glucopyranoside (octyl glucoside, OG, Sigma). αIIbβ3 concentrations were determined by ultraviolet absorbance measurements (LKB Ultrospec) using an extinction coefficient at 280 m of 1.21 mL mg−1 cm−1, as previously described (Ramsamooj et al. 1990; Hantgan et al. 1993). Samples of αIIbβ3 obtained prior to and following exposure to ligand-mimetic or control peptides were analyzed by SDS-PAGE using 5%–15% gradient gels (Bio Rad) and followed by protein staining with Coomassie Brilliant Blue R-250, as previously described (Hantgan et al. 1999). Selected samples were electrophoretically transferred to PVDF membranes and immunostained with monoclonal antibodies specific for either the αIIb or β3 subunits (CD61 and CD41, respectively, Immunotech). Immune complexes were detected with an alkaline phosphatase-conjugated goat anti-mouse monoclonal antibody using a BioRad Enhanced Chemiluminescence kit with images captured on film for subsequent analyses using Sigma Gel software (Jandel Scientific).
Ligand-mimetic and control peptides
Eptifibatide, (N6– (aminoiminomethyl)-N2– (3-mercapto-1-oxopropyl - L - lysylglycyl - L - α-aspartyl - L - tryptophanyl - L - prolyl -cysteinamide, cyclic (1–6)-disulfide), was provided by COR Therapeutics (San Francisco, CA), where it has been developed as a pharmaceutical, Integrilin (Phillips and Scarborough 1997). An inactive control peptide, cyclo-L-cysteinyl-L-lysyl-D-alanyl-L-aspartyl-L-tryptophanyl-L-prolyl-L-cystinyl-amide (CKADWPC), was also provided by COR Therapeutics. Both peptide preparations were dry powders that were dissolved in HSC-OG and stored for up to 6 months at −70°C. Peptide concentrations were determined by quantitative amino acid composition analyses (Hantgan et al. 1992) of samples of stock solutions.
Static and dynamic light-scattering instrumentation and data analyses
The molecular weight and translational diffusion coefficient of the αIIbβ3 complex were measured with a Brookhaven Instruments BI-2030 AT correlator, operated in conjunction with a BI-200 SM light scattering goniometer/photon counting detector and a Spectra Physics 127 He-Ne laser (35 mW, equipped with a vertical polarization rotator), as previously described (Hantgan et al. 1993, 1999).
For molecular weight determinations, right-angle sample scattering intensities were corrected for solvent scattering and expressed relative to a benzene standard (Hantgan et al. 1993). This information, coupled with measurements of protein concentration by UV-absorbance, was used to calculate the weight-average molecular weight (Mw) of αIIbβ3 samples using Rayleigh-Gans theory (Johnson and Gabriel 1981). For translational diffusion coefficient determinations, each intensity-normalized photon count autocorrelation function obtained for the αIIbβ3 complex was corrected for the contributions of octyl glucoside micelles and then analyzed by the method of cumulants, as previously described (Hantgan et al. 1993). All translational diffusion coefficients reported here have been corrected for solvent viscosity to obtain D20,w values.
Analytical ultracentrifugation: Instrumentation and data analyses
Sedimentation velocity and equilibrium measurements were performed in a Beckman Optima XL-A analytical ultracentrifuge (Beckman Instruments) equipped with absorbance optics and an An60 Ti rotor. Sedimentation velocity data for the αIIbβ3 complex (alone and in the presence of ligand-mimetic or control peptide) were obtained in double-sector cells at 20°C at a rotor speed of 35,000 rpm, as previously described (Hantgan et al. 1999). These data were analyzed using both SVEDBERG (version 1.04) and DCDT+ (version 6.31) software (J. Philo) to obtain the weight-average sedimentation coefficient (Sw) and distribution of sedimenting species, g (s*), respectively (Stafford III 1992). All sedimentation coefficients reported here have been corrected for solvent density and viscosity to obtain S20, w values.
Sedimentation equilibrium data were collected from αIIbβ3 samples (alone and in the presence of ligand-mimetic or control peptide) contained in double-sector cells with buffered octyl glucoside (and peptides, as required) in the reference sector. Data were collected at 280 nm at rotor speeds of 6000 and 8000 rpm at 20°C, as previously described (Hantgan et al. 1999). The absorbance versus radial distance data were analyzed by nonlinear regression with WinNONLIN3 (Johnson et al. 1981) to obtain the self-association constant for the αIIbβ3 complex alone and in the presence of ligand-mimetic (or control) peptide. WinNONLIN3 was provided by David Yphantis and the staff at the National Analytical Ultracentrifugation Facility.
Spectroscopic measurements
UV absorbance measurements of αIIbβ3 samples contained in 1-cm path length/1 mL volume quartz cuvettes were performed as a function of wavelength on a Milton Roy Spectronic 3000 Array spectrophotometer (chosen for its 0.3-nm resolution); single wavelength determinations were performed on an LKB Uvicord II instrument. Fluorescence emission spectra were obtained with an AMINCO-Bowman Series 2 Luminescence Spectrometer (SLM-AMINCO). Samples were excited at 278 nm (1-nm bandwidth) and the emission spectra recorded from 300–500 nm (1-nm bandwidth). Samples were contained within quartz microcuvettes (1-cm pathlength, 150 μL filling volume, 4 windows, Hellma Cells, Inc.). All spectral measurements were obtained at 23 ± 1°C and corrected for the contributions of octyl glucoside buffer, eptifibatide, and guanidinium chloride, when present in the samples.
Rotary-shadowed specimens for electron microscopy
Rotary-shadowed samples were prepared by spraying a dilute solution (final concentration ∼20–25 μg/mL) of molecules in a volatile buffer, 0.05 M ammonium formate at pH 7.4, 30 mM octyl glucoside, and 30% (v/v) glycerol onto freshly cleaved mica and shadowing with tungsten in a vacuum evaporator (Denton Vacuum Co.; Fowler and Erickson 1979; Weisel et al. 1985; Veklich et al. 1993). These samples were examined in a Philips CM100 electron microscope (FEI Co.) operating at 60 kV and a magnification of 53,000×. Counts of molecules with different conformations or different amounts of oligomers were made from prints of the micrographs, using images from many different areas of several different preparations to get a random sample.
Molecular modeling
Bead models of the αIIbβ3 complex (Hantgan et al. 1999) were constructed, visualized, and analyzed using the BEAMS (BEAds Modeling System) set of computer programs to represent each of the αIIb and β3 polypeptide chains of the αIIbβ3 complex as an ensemble of interconnected spheres (Rocco et al. 1993; Spotorno et al. 1997; see also Byron 2000). Hydrodynamic theory for multisubunit particles (De La Torre and Bloomfield 1981; Spotorno et al. 1997;) was employed to calculate the translational diffusion coefficient, Dt, the Stokes Radius, Rs, and the sedimentation coefficient, s, for the models as previously described (Rocco et al. 1993; Hantgan et al. 1999). Molecular models of eptifibatide and CKDAWPC were constructed and a minimum energy configuration for each obtained (Cohen et al. 1990) using ALCHEMY 2000 and SYBYL software (Tripos).
Acknowledgments
Thanks to Drs. R.M. Scarborough, D.R. Phillips, M.M. Kitt, and S. Hollenbach of COR Therapeutics, Inc., for providing eptifibatide and the control cyclic peptide, as well as for helpful discussions; Mary Stahle for her expert technical assistance; and Dr. D.S. Lyles for his critical reading of the manuscript and to Dr. J.B. Edelson for her skilled editing. Support was provided by grant MCB-9728122 from the National Science Foundation (to R.R.H.), grant BI-04-CT96–0662 from the European Community (to M.R.), and grant HL30954 from the National Institutes of Health (to J.W.W.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.3001
References
- Basani, R.B., D'Andrea, G., Mitra, N., Vilaire, G., Richberg, M., Kowalska, M.A., Bennett, J.S., and Poncz, M. 2001. RGD-containing peptides inhibit fibrinogen binding to platelet αIIbβ3 by inducing an allosteric change in the amino-terminal portion of αIIb. J. Biol. Chem. 276 13975–13981. [DOI] [PubMed] [Google Scholar]
- Bu, Z. and Engelman, D.M. 1999. A method for determining transmembrane helix association and orientation in detergent micelles using small angle x-ray scattering. Biophys. J. 77 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron, O. 2000. Hydrodynamic bead modeling of biological macromolecules. Meth. Enzymol. 321 278–304. [DOI] [PubMed] [Google Scholar]
- Cantor, C.R. and Schimmel, P.R. 1980. Other optical techniques. In Biophysical Chemistry. Part II: Techniques for the study of biological structure and function, pp. 409–480. WH Freeman and Co., San Francisco.
- Carrell, N.A., Fitzgerald, L.A., Steiner, B., Erickson, H.P., and Phillips, D.R. 1985. Structure of human platelet glycoproteins IIb and IIIa as determined by electron microscopy. J. Biol. Chem. 260 1743–1749. [PubMed] [Google Scholar]
- Chiu, C.Y., Mathias, P., Nemerow, G.R., and Stewart, P.L. 1999. Structure of adenovirus complexed with its internalization receptor, αvβ5 integrin. J. Virol. 73 6759–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cierniewski, C.S., Byzova, T., Papierak, M., Haas, T.A., Niewiarowska, J., Zhang, L., Cieslak, M., and Plow, E.F. 1999. Peptide ligands can bind to distinct sites in integrin αIIbβ3 and elicit different functional responses. J. Biol. Chem. 274 16923–16932. [DOI] [PubMed] [Google Scholar]
- Clark, E.A. and Brugge, J.S. 1995. Integrins and signal transduction pathways: The road taken. Science 268 233–239. [DOI] [PubMed] [Google Scholar]
- Cohen, N.C., Blaney, J.M., Humblet, C., Gund, P., and Barry, D.C. 1990. Molecular modeling software and methods for medicinal chemistry. J. Med. Chem. 33 883–894. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1984. Proteins in Solution and in Membranes. In Proteins: Structures and Molecular Properties, (ed. T.E. Creighton), pp. 261–328. WH Freeman and Co., New York.
- Critchley, D.R., Holt, M.R., Barry, S.T., Priddle, H., Hemmings, L., and Norman, J. 1999. Integrin-mediated cell adhesion: The cytoskeletal connection. Biochem. Soc. Symp. 65 79–99. [PubMed] [Google Scholar]
- D'Souza, S.E., Ginsberg, M.H., Burke, T.A., Lam, S.C.T., and Plow, E.F. 1988. Localization of an arg-gly-asp recognition site within an integrin adhesion receptor. Science 242 91–93. [DOI] [PubMed] [Google Scholar]
- D'Souza, S.E., Ginsberg, M.H., Burke, T.A., and Plow, E.F. 1990. The ligand binding site of the platelet integrin receptor GPIIb-IIIa is proximal to the second calcium binding domain of its α subunit. J. Biol. Chem. 265 3440–3446. [PubMed] [Google Scholar]
- De La Torre, J.G. and Bloomfield, V.A. 1981. Hydrodynamic properties of complex, rigid biological macromolecules: Theory and applications. Quart. Rev. Biophys. 14 81–139. [DOI] [PubMed] [Google Scholar]
- Dedhar, S. and Hannigan, G.E. 1996. Integrin cytoplasmic interactions and bidirectional transmembrane signalling. Curr. Opin. Cell Biol. 8 657–669. [DOI] [PubMed] [Google Scholar]
- Dickeson, S.K. and Santoro, S.A. 1998. Ligand recognition by the I domain-containing integrins. Cell. Mol. Life Sci. 54 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue, J.P., Patel, H., Anderson, W.F., and Hawiger, J. 1994. Three-dimensional structure of the platelet integrin recognition segment of the fibrinogen (chain obtained by carrier protein-driven crystallization. Proc. Natl. Acad. Sci. 91 12178–12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, X., Gu, M., Weisel, J.W., Nagaswami, C., Bennett, J.S., Bowditch, R., and Ginsberg, M.H. 1993. Long range propagation of conformational changes in integrin αIIbβ3. J. Biol. Chem. 268 23087–23092. [PubMed] [Google Scholar]
- Emsley, J., Knight, C.G., Farndale, R.W., Barnes, M.J., and Liddington, R.C. 2000. Structural basis of collagen recognition by integrin α2β1. Cell 101 47–56. [DOI] [PubMed] [Google Scholar]
- Erb, E.M., Tangemann, K., Bohrmann, B., Müller, B., and Engel, J. 1997. Integrin αIIbβ3 reconstituted into lipid bilayers is nonclustered in its activated state but clusters after fibrinogen binding. Biochemistry 36 7395–7402. [DOI] [PubMed] [Google Scholar]
- Farrell, D.H., Thiagarajan, P., Chung, D.W., and Davie, E.W. 1992. Role of fibrinogen α and γ chain sites in platelet aggregation. Proc. Natl. Acad. Sci. 89 10729–10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald, L.A., Steiner, B., Rall, S.C., Lo, S., and Phillips, D.R. 1987. Protein sequence of endothelial glycoprotein IIIa derived from a cDNA clone. J. Biol. Chem. 262 3936–3939. [PubMed] [Google Scholar]
- Fowler, W.E. and Erickson, H.P. 1979. Trinodular structure of fibrinogen. Confirmation by both shadowing and negative stain electron microscopy. J. Mol.Biol. 134 241–249. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G. and Ruoslahti, E. 1999. Integrin signaling. Science 285 1028–1032. [DOI] [PubMed] [Google Scholar]
- Ginsberg, M.H., O'Toole, T.E., Loftus, J.C., and Plow, E.F. 1992. Ligand binding to integrins: Dynamic regulation and common mechanisms. Cold Spring Harbor Symposia on Quantitative Biology 57 221–231. [DOI] [PubMed] [Google Scholar]
- Goa, K.L. and Noble, S. 1999. Eptifibatide—A review of its use in patients with acute coronary syndromes and/or undergoing percutaneous coronary intervention. Drugs 57 439–462. [DOI] [PubMed] [Google Scholar]
- Haas, T.A. and Plow, E.F. 1997. Development of a structural model for the cytoplasmic domain of an integrin. Protein Eng. 10 1395–1405. [DOI] [PubMed] [Google Scholar]
- Hantgan, R.R., Braaten, J.V., and Rocco, M. 1993. Dynamic light scattering studies of αIIbβ3 solution conformation. Biochemistry 32 3935–3941. [DOI] [PubMed] [Google Scholar]
- Hantgan, R.R., Endenburg, S.C., Cavero, I., Marguerie, G., Uzan, A., Sixma, J.J., and De Groot, P.G. 1992. Inhibition of platelet adhesion to fibrin(ogen) in flowing whole blood by Arg-Gly-Asp and fibrinogen gamma-chain carboxy terminal peptides. Thromb .Haemost. 68 694–700. [PubMed] [Google Scholar]
- Hantgan, R.R., Paumi, C., Rocco, M., and Weisel, J.W. 1999. Effects of ligand-mimetic peptides Arg-Gly-Asp-X (X = Phe, Trp, Ser) on αIibβ3 integrin conformation and oligomerization. Biochemistry 38 14461–14474. [DOI] [PubMed] [Google Scholar]
- Hawiger, J. 1995. Adhesive ends of fibrinogen and its antiadhesive peptides: The end of a saga. Semin. Hematol. 32 99–109. [PubMed] [Google Scholar]
- Hawiger, J., Kloczewiak, M., Bednarek, M.A., and Timmons, S. 1989. Platelet receptor recognition domains on the α chain of human fibrinogen: Structure-function analysis. Biochemistry 28 2909–2914. [DOI] [PubMed] [Google Scholar]
- Hettasch, J.M., Bolyard, M.G., and Lord, S.T. 1992. The residues AGDV of recombinant gamma chains of human fibrinogen must be carboxy-terminal to support human platelet aggregation. Thromb. Haemost. 68 701–706. [PubMed] [Google Scholar]
- Hu, D.D., White, C.A., Panzer-Knodle, S., Page, J.D., Nicholson, N., and Smith, J.W. 1999. A new model of dual interacting ligand binding sites on integrin αIIbβ3. J. Biol.Chem. 274 4633–4639. [DOI] [PubMed] [Google Scholar]
- Huang, C.C. and Springer, T.A. 1997. Folding of the β-propeller domain of the integrin αL subunit is independent of the I domain and dependent on the β2 subunit. Proc.Natl.Acad.Sci.USA 94 3162–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, P.E. and Pfaff, M. 1998. Integrin affinity modulation. TICB 8 359–364. [DOI] [PubMed] [Google Scholar]
- Humphries, M.J. 1999. Towards a structural model of an integrin. Biochemical Society Symposia 65 63–78. [PubMed] [Google Scholar]
- Hynes, R.O. 1992. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 69 11–25. [DOI] [PubMed] [Google Scholar]
- Johnson, C.S. and Gabriel, D.A. 1981. Laser light scattering. In Spectroscopy in Biochemistry. Vol. II (ed J.E. Bell), pp. 177–272. CRC Press, Boca Raton, Florida.
- Johnson, M., Correia, J.J., Yphantis, D.A., and Halvorson, H. 1981. Analysis of data from the analytical ultracentrifuge by nonlinear leastsquare techniques. Biophys..J. 36 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloczewiak, M., Timmons, S., Bednarek, M.A., Sakon, M., and Hawiger, J. 1989. Platelet receptor recognition domain on the gamma chain of human fibrinogen and its synthetic peptide analogues. Biochemistry 28 2915–2919. [DOI] [PubMed] [Google Scholar]
- Koshland, D.E.J. 1998. Conformational changes: How small is big enough?: Nat. Med. 4 1112–1114. [DOI] [PubMed] [Google Scholar]
- Leahy, D.J. 1997. Implications of atomic-resolution structures for cell adhesion. Annu. Rev. Cell Dev. Biol. 13 363–393. [DOI] [PubMed] [Google Scholar]
- Lee, J.-O., Rieu, P., Arnaout, M.A., and Liddington, R. 1995. Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18). Cell 80 631–638. [DOI] [PubMed] [Google Scholar]
- Lee, J.O., Bankston, L.A., Arnaout, M.A., and Liddington, R.C. 1995. Two conformations of the integrin A-domain (I-domain): A pathway for activation? Structure 3 1333–1340. [DOI] [PubMed] [Google Scholar]
- Leisner, T.M., Wencel-Drake, J.D., Wang, W., and Lam, S.C. 1999. Bidirectional transmembrane modulation of integrin αIIbβ3 conformations. J. Biol. Chem. 274 12945–12949. [DOI] [PubMed] [Google Scholar]
- Leitinger, B. and Hogg, N. 2000. From crystal clear ligand binding to designer I domains. Nat. Struct. Biol. 7 614–616. [DOI] [PubMed] [Google Scholar]
- Liddington, R.C. and Bankston, L.A. 2000. The structural basis of dynamic cell adhesion: Heads, tails, and allostery. Exper. Cell Res. 261 37–43. [DOI] [PubMed] [Google Scholar]
- Loftus, J.C. and Liddington, R.C. 1997. New insights into integrin-ligand interaction. J. Clin. Invest. 99 2302–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber, B., Bishop, J.B., and DeLucas, L.J. 1990. Purification of octyl beta-D-glucopyranoside and re-estimation of its micellar size. Biochimica et Biophysica Acta 1023 254–265. [DOI] [PubMed] [Google Scholar]
- MacKenzie, K.R., Prestegard, J.H., and Engelman, D.M. 1997. A transmembrane helix dimer: Structure and implications. Science 276 131–133. [DOI] [PubMed] [Google Scholar]
- Minoux, H., Chipot, C., Brown, D., and Maigret, B. 2000. Structural analysis of the KGD sequence loop of barbourin, an alpha(IIb)beta(3)-specific disintegrin. J. Comp.-Aided Mol. Design 14 317–327. [DOI] [PubMed] [Google Scholar]
- Minoux, H., Moitessier, N., Chapleur, Y., and Maigret, B. 1998. Elucidation of a common structure of selective fibrinogen receptor antagonists. J. Comp.-Aided Mol. Design 12 533–542. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S., Akiyama, S.K., and Yamada, K.M. 1995. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science 267 883–885. [DOI] [PubMed] [Google Scholar]
- O'Toole, T.E., Katagiri, Y., Faull, R.J., Peter, K., Tamura, R., Quaranta, V., Loftus, J.C., Shattil, S.J., and Ginsberg, M.H. 1994. Integrin cytoplasmic domains mediate inside-out signal transduction. J.Cell Biol. 124 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottemann, K.M., Xiao, W., Shin, Y.K., and Koshland, D.E.J. 1999. A piston model for transmembrane signaling of the aspartate receptor. Science 285 1751–1754. [DOI] [PubMed] [Google Scholar]
- Parise, L.V., Helgerson, S.L., Steiner, B., Nannizzi, L., and Phillips, D.R. 1987. Synthetic peptides derived from fibrinogen and fibronectin change the conformation of purified glycoprotein IIb-IIIa. J.Biol.Chem. 262 12597–12602. [PubMed] [Google Scholar]
- Peterson, J.A., Visentin, G.P., Newman, P.J., and Aster, R.H. 1998. A recombinant soluble form of the integrin αIIbβ3 (GpIIb-IIIa) Assumes an active, ligand-binding conformation and is recognized by gpIIb-IIIa specific monoclonal, allo-, auto-, and drug-dependent platelet antibodies. Blood 92 2053–2063. [PubMed] [Google Scholar]
- Phillips, D.R., Charo, I.F., Parise, L.V., and Fitzgerald, L.A. 1988. The platelet membrane glycoprotein IIb-IIIa complex. Blood 71 831–843. [PubMed] [Google Scholar]
- Phillips, D.R. and Scarborough, R.M. 1997. Clinical pharmacology of eptifibatide. Am. J. Cardiol. 80 11B–20B. [DOI] [PubMed] [Google Scholar]
- Plow, E.F., Haas, T.K., Zhang, L., Loftus, J., and Smith, J.W. 2000. Ligand binding to integrins. J. Biol. Chem. 275 21785–21788. [DOI] [PubMed] [Google Scholar]
- Plow, E.F., Pierschbacher, M.D., Ruoslahti, E., Marguerie, G., and Ginsberg, M.H. 1985. The effect of arg-gly-asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets. Proc. Natl. Acad. Sci. 82 8057–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncz, M., Eisman, R., Heidenreich, R., Silver, S.M., Vilaire, G., Surrey, S., Schwartz, E., and Bennett, J.S. 1987. Structure of the platelet membrane glycoprotein IIb. Homology to the A subunits of the vitronectin and fibronectin membrane receptors. J. Biol. Chem. 262 8476–8482. [PubMed] [Google Scholar]
- Ramsamooj, P., Doellgast, G.J., and Hantgan, R.R. 1990. Inhibition of fibrin(ogen) binding to stimulated platelets by a monoclonal antibody specific for a conformational determinant of gpIIIa. Thromb. Res. 58 577–592. [DOI] [PubMed] [Google Scholar]
- Rocco, M., Spotorno, B., and Hantgan, R.R. 1993. Modeling the αIIbβ3 integrin solution conformation. Prot. Sci. 2 2154–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti, E. and Pierschbacher, M.D. 1987. New perspectives in cell adhesion: RGD and integrins. Science 238 491–497. [DOI] [PubMed] [Google Scholar]
- Santoro, S.A. and Lawing Jr., W.J. 1987. Competition for related but nonidentical binding sites on the glycoprotein IIb-IIIa complex by peptides derived from platelet adhesive proteins. Cell 48 867–873. [DOI] [PubMed] [Google Scholar]
- Scarborough, R.M., Rose, J.W., Hsu, M.A., Phillips, D.R., Fried, V.A., Campbell, A.M., Nannizzi, L., and Charo, I.F. 1991. Barbourin. A GPIIb-IIIa-specific integrin antagonist from the venom of Sistrurus m. barbouri. J. Biol. Chem. 266 9359–9362. [PubMed] [Google Scholar]
- Scarborough, R.M., Naughton, M.A., Teng, W., Rose, J.W., Phillips, D.R., Nannizzi, L., Arfsten, A., Campbell, A.M., and Charo, I.F. 1993a. Design of potent and specific integrin antagonists. Peptide antagonists with a high specificity for glycoprotein IIb-IIIa. J. Biol. Chem. 268 1066–1073. [PubMed] [Google Scholar]
- Scarborough, R.M., Rose, J.W., Naughton, M.A., Phillips, D.R., Nannizzi, L., Arfsten, A., Campbell, A.M., and Charo, I.F. 1993b. Characterization of the integrin specificities of disintegrins isolated from American pit viper venoms. J. Biol. Chem. 268 1058–1065. [PubMed] [Google Scholar]
- Shattil, S.J. 1999. Signaling through platelet integrin αIIbβ3: Inside-out, outside-in, and sideways. Thromb. Haemost. 82 318–325. [PubMed] [Google Scholar]
- Shattil, S.J., Kashiwagi, H., and Pampori, N. 1998. Integrin signaling: The platelet paradigm. Blood 91 2645–2657. [PubMed] [Google Scholar]
- Sims, P.J., Ginsberg, M.H., Plow, E.F., and Shattil, S.J. 1991. Effect of platelet activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J. Biol. Chem. 266 7345–7352. [PubMed] [Google Scholar]
- Spotorno, B., Piccinini, L., Tassara, G., Ruggiero, C., Nardini, M., Molina, F., and Rocco, M. 1997. BEAMS (BEAds Modeling System): A set of computer programs for the visualization and the computation of hydrodynamic and conformational properties of beads models of proteins. Eur. Biophys. J. 25 373–384. [Google Scholar]
- Springer, T.A. 1997. Folding of the N-terminal, ligand-binding region of integrin α-subunits into a β-propeller domain. Proc. Natl. Acad. Sci. 94 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford III, W.F. 1992. Boundary analysis in sedimentation transport experiments: A procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 203 295–301. [DOI] [PubMed] [Google Scholar]
- Suehiro, K. and Plow, E.F. 1997. Ligand recognition by beta 3 integrins. Keio J. Med. 46 111–114. [DOI] [PubMed] [Google Scholar]
- Thibault, G. 2000. Sodium dodecyl sulfate-stable complexes of echistatin and RGD-dependent integrins: A novel approach to study integrins. Mol. Pharmacol. 58 1137–1145. [DOI] [PubMed] [Google Scholar]
- VanAken, T., Foxall-VanAken, S., Castleman, S., and Ferguson-Miller, S. 1986. Alkyl glycoside detergents: Synthesis and applications to the study of membrane proteins. Meth. Enzymol. 125 27–35. [DOI] [PubMed] [Google Scholar]
- Veklich, Y.I., Gorkun, O.V., Medved, L.V., Nieuwenhuizen, W., and Weisel, J.W. 1993. Carboxyl-terminal portions of the α chains of fibrinogen and fibrin. Localization by electron microscopy and the effects of isolated αC fragments on polymerization. J. Biol. Chem. 268 13577–13585. [PubMed] [Google Scholar]
- Vinogradova, O., Haas, T., Plow, E.F., and Qin, J. 2000. A structural basis for integrin activation by the cytoplasmic tail of the αIIb-subunit. Proc. Natl. Acad. Sci. 97 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware, S., Donahue, J.P., Hawiger, J., and Anderson, W.F. 1999. Structure of the fibrinogen gamma-chain integrin binding and factor XIIIa cross-linking sites obtained through carrier protein driven crystallization. Protein Sci. 8 2663–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel, J.W., Nagaswami, C., Vilaire, G., and Bennett, J.S. 1992. Examination of the platelet membrane glycoprotein IIb-IIIa complex and its interaction with fibrinogen and other ligands by electron microscopy. J. Biol. Chem. 267 16637–16643. [PubMed] [Google Scholar]
- Weisel, J.W., Stauffacher, C.V., Bullitt, E., and Cohen, C. 1985. A model for fibrinogen: Domains and sequence. Science 230 1388–1391. [DOI] [PubMed] [Google Scholar]
- Wells, J.A. and deVos, A.M. 1996. Hematopoietic Receptor Complexes. Annu. Rev. Biochem. 65 609–634. [DOI] [PubMed] [Google Scholar]
- Wippler, J., Kouns, W.C., Schlaeger, E.-J., Kuhn, H., Hadvary, P., and Steiner, B. 1994. The integrin αIIb-β3, platelet glycoprotein IIb-IIIa, can form a functionally active heterodimer complex without the cysteine-rich repeats of the β3 subunit. J. Biol. Chem. 269 8754–8761. [PubMed] [Google Scholar]
- Yan, B. and Smith, J.W. 2000. A Redox Site Involved in Integrin Activation. J. Biol. Chem. 275 39964–39972. [DOI] [PubMed] [Google Scholar]
- Yee, V.C., Pratt, K.P., Côté, H.C.F., Le Trong, I., Chung, D.W., Davie, E.W., Stenkamp, R.E., and Teller, D.C. 1997. Crystal structure of a 30 kDa C-terminal fragment from the gamma chain of human fibrinogen. Structure 5 125–138. [DOI] [PubMed] [Google Scholar]