

Abstract
The design of safe sweeteners is very important for people who are affected by diabetes, hyperlipemia, and caries and other diseases that are linked to the consumption of sugars. Sweet proteins, which are found in several tropical plants, are many times sweeter than sucrose on a molar basis. A good understanding of their structure–function relationship can complement traditional SAR studies on small molecular weight sweeteners and thus help in the design of safe sweeteners. However, there is virtually no sequence homology and very little structural similarity among known sweet proteins. Studies on mutants of monellin, the best characterized of sweet proteins, proved not decisive in the localization of the main interaction points of monellin with its receptor. Accordingly, we resorted to an unbiased approach to restrict the search of likely areas of interaction on the surface of a typical sweet protein. It has been recently shown that an accurate survey of the surface of proteins by appropriate paramagnetic probes may locate interaction points on protein surface. Here we report the survey of the surface of MNEI, a single chain monellin, by means of a paramagnetic probe, and a direct assessment of bound water based on an application of ePHOGSY, an NMR experiment that is ideally suited to detect interactions of small ligands to a protein. Detailed surface mapping reveals the presence, on the surface of MNEI, of interaction points that include residues previously predicted by ELISA tests and by mutagenesis.
Keywords: Monellin, sweet proteins, taste, NMR structure, surface accessibility, hydration
The use of low-calorie sweeteners is of paramount importance for people affected by diseases, such as diabetes, hyperlipemia, and caries, that are more or less directly linked to the assumption of sugar. The design of safe sweeteners relies upon an in-depth understanding of the SAR of sweet molecules and of their interaction with the receptor. Most sweeteners are small molecular weight compounds (Moncrieff 1967), but there are also sweet macromolecules, both synthetic (Zaffaroni 1975) and natural (i.e., sweet proteins [Morris 1976]). Considering the enormous difference in size among low-molecular-weight sweeteners and sweet proteins, and even among proteins, it has often been doubted that these two classes interact with the same receptor. However, both immunochemical studies and models of the receptor active site hint that all sweet molecules probably interact with the same receptor. ELISA tests showed cross-reactivity among antibodies raised against monellin (94 residues) with thaumatin (210 residues) and aspartame (2 residues), and hinted that the sequence TyrA13-AspA16 of native monellin, which has its counterpart in residues Tyr57-Asp59 of thaumatin, is a potential "sweet finger" (i.e., protruding structural elements that host glucophores) (Kim et al. 1991).
The key groups on the protein surface that are responsible for the biological activity have not yet been identified with certainty for any of the known sweet proteins (Caldwell et al. 1998). However, if we assume that the glucophores on the protein surface are similar to those that account for the taste of small molecules, it is possible to hypothesize the presence, on the protein surface, of "sweet fingers." Models of receptor active site (Shallenberger and Acree 1967; Kier 1972; Temussi et al. 1978, 1984, 1991; Iwamura 1981; Goodman et al. 1987) contain, as main glucophores, a hydrogen bond donor (AH) and a hydrogen bond acceptor (B) spaced 3 Å–4 Å but differ for stereochemical aspects. Our model for the sweet receptor (Temussi et al. 1991) is consistent also with macromolecules, given that the active site is depicted as an open cavity with a flat bottom that is accessible to a sweet finger. However, none of the existing models for the sweet receptor can explain the enormous increase in activity in going from small molecular weight compounds to proteins; for example monellin, one of the best characterized sweet proteins, is 100,000 times sweeter than sucrose on a molar basis (Hung et al. 1999).
If one has several proteins with the same function, it is customary to try to identify corresponding parts, responsible for the interaction with a receptor, by examining their sequences or their 3D structures. However, there is virtually no sequence homology among miraculin (Kurihara and Beidler 1968), monellin (Inglett and May 1969), thaumatin (van der Wel and Loeve 1972), curculin (Harada et al. 1994), mabinlin (Liu et al. 1993), and brazzein (Ming and Hellekant 1994) and very little structural similarity among the 3D structures of monellin, thaumatin, and brazzein, as calculated by DALI (Holm and Sander 1995). Another commonly used strategy is to mutate systematically all residues that are suspected to host potential glucophores or that have a key structural role. Extensive mutagenesis studies on monellin (Ariyoshi and Kohmura 1994) and on single chain monellin (SCM), one of its single chain mutants (Somoza et al. 1995), although yielding several constructs with substantial loss of activity, are not decisive in the localization of the main interaction points of monellin with the receptor because the results are not fully self consistent and, to some extent, contradict previous studies based on ELISA tests (Kim et al. 1991).
Considering these difficulties, we resorted to an unbiased approach to restrict the search of likely areas of interaction on the surface of a typical sweet protein. Several studies, carried out both in solution (Liepinsh and Otting 1997; Dalvit et al. 1999) and in the crystal state (Ringe 1995; Mattos and Ringe 1996), have recently pointed out that proteins have regions where small and uncharged organic molecules, even different from their physiological ligands, preferentially approach the molecular surface. These "hot spots" of the protein surface account for the disruption of substrate binding at sites different from the expected ones (Krantz 1998; Savvides and Karplus 1996). It has been recently proposed that an accurate survey of the surface of proteins by appropriate paramagnetic probes may help to locate these interaction points that is, it can reveal hot spots on the protein surface (Scarselli et al. 1999). This behavior is very general and can be ascribed to a reduced strength in the competitive binding of water molecules to these regions. If one takes into account the fact that regions exhibiting such propensities always include the protein active site, it is possible to envisage a sort of vortex effect that contributes to the efficiency and precision of ligand binding.
The sweet protein we chose for the experimental study is MNEI, a construct of 96 amino acid residues engineered by linking, with a Gly–Phe dipeptide, chains B and A of monellin (Tancredi et al. 1992). Monellin, a protein that in its natural form is composed of two chains, A and B, linked by noncovalent interactions, is the best characterized of sweet proteins from a structural point of view. Single chain mutants, that is, monellins in which the two chains are covalently linked, can retain all sweetening power but have greatly increased thermal stability (Kim et al. 1989; Tancredi et al. 1992). The solution structure of MNEI, recently solved by us (Spadaccini et al. 2001), is very similar to the solid-state structures of both native monellin and SCM and can thus be used to examine also point mutations of native monellin and of SCM. Here we report the surface mapping of MNEI to assess TEMPOL and water accessibility. The paramagnetic probing is complemented by a direct assessment of bound water based on an application of ePHOGSY, an NMR experiment ideally suited to detect interactions of small ligands to a protein.
Results
Resonance assignments and TEMPOL attenuation
Spectra of MNEI alone or in the presence of suitable amounts of TEMPOL were recorded in solution conditions (pH 2.9,35°C) identical to those employed in the structure determination (Spadaccini et al. 2001). Chemical shifts are only marginally affected by the presence of TEMPOL (mean absolute difference <0.02 ppm), a circumstance that makes assignments trivial. Resonance assignments were nonetheless accurately checked by standard procedures based on COSY (Aue et al. 1976), TOCSY (Bax and Davis 1985) and NOESY (Jeener et al. 1979) experiments.
A 16 mM TEMPOL concentration, although inducing a sizeable broadening of MNEI proton signals, does not generate too much loss in the signal/noise (S/N) ratio. Accordingly, all attenuation data that are reported here refer to this experimental condition. Because denaturing effects of TEMPOL on the protein could not be excluded a priori, after each addition of the 1M solution of TEMPOL to the 2 mM MNEI solution, 1D MNEI spectra were recorded to check for spectral modifications. Thus, at the final 16 mM concentration of the paramagnetic probe, all residues exhibit chemical shift changes <0.018 ppm with the exception of D7(NH), V37(NH), I46(NH), Y58(NH), S76(HB1), and R88(HG1, HZ), whose changes range between 0.04 ppm and 0.06 ppm.
TOCSY spectra of MNEI show a large variety of well-resolved cross-peaks, and it is easy to identify many homologous J connectivities from the different types of residues. The aliphatic region of the TOCSY map of MNEI, recorded in diamagnetic and paramagnetic water solutions, is shown in Figure 1A ▶ and Figure 1B ▶, respectively. A decreased number of scalar connectivities in the presence of TEMPOL is readily seen. Intensities of cross-peaks are selectively affected, as illustrated by Figure 1C ▶ and Figure 1D ▶, which show a blow up of a smaller region containing the α–β cross-peaks of Asp68. To reduce possible ambiguities in the quantitative analysis of the NMR data, only the paramagnetic attenuations of nonoverlapping 2D cross-peaks were considered. In the case of nonequivalent methylene protons, mean attenuations have been reported.
Fig. 1.

Comparison of the aliphatic regions of the TOCSY of MNEI, recorded in diamagnetic (A) and paramagnetic (B) water solutions. NMR spectra were recorded on 2.0 mM samples in an H2O/2H2O (90:10, v.v) solution containing 18.5 mM potassium phosphate, adjusted to pH 2.9, at 35°C. The paramagnetic samples are also 16 mM in TEMPOL. Panels (C) and (D) show a blow-up of a smaller region containing the strongly attenuated α–β cross-peak of D68.
Quantitative evaluation of TEMPOL accessibility was performed as previously described, by measuring autoscaled cross-peak attenuation figures Ai (Molinari et al. 1997). A detailed description of the calculations is illustrated in Materials and Methods (vide infra). The most reliable quantitative information, as far as surface properties are concerned, in particular the identification of possible interaction points, can be gathered from α–β J connectivities because they are least affected by phenomena connected with exchange of labile protons (e.g., direct hydrogen bonding to TEMPOL). Another useful feature of these correlations is the fact that connectivities of protons more distant from the backbone may reflect local mobility, whereas paramagnetic attenuations of α–β J connectivities are expected to undergo reorientation at a rate reflecting the overall protein motion. Attenuations of α–β J connectivities corresponding to well-resolved resonances are shown in Figure 2 ▶.
Fig. 2.

Histogram of attenuations and accessibility. The autoscaled paramagnetic cross-peak attenuations Ai, calculated for all the well resolved correlations of MNEI residues in (A) TOCSY and (B) ePHOGSY–TOCSY spectra. Black bars refer to static accessibility calculated as described in the text.
For a better assessment of paramagnetic effects, Ai values can be profitably compared to exposed surface areas. Exposed surface areas, ESAi, have been calculated with the program MOLMOL (Koradi et al. 1996) a probe radius of 1.4 Å corresponding to water molecules, using the MNEI hydrogen coordinates of the NMR derived structure of the Protein Data Bank (PDB name 1fa3; Spadaccini et al. 2001). When considering the intensity variations of a TOCSY cross-peak in response to the paramagnetic perturbation, a different contribution from each of the two protons that originate a given J connectivity in the cross-peak intensity should be expected in principle. However, in agreement with previous suggestions (Molinari et al. 1997), only the sum of the ESAis of the nuclei that originate a J connectivity is considered in this report and used to discuss the correlation of the computed surface exposure with the paramagnetic attenuations.
The residues whose α–β J connectivities are attenuated by TEMPOL are spread all over the sequence of MNEI. If we restrict our attention to Ai values >1.4, the residues most affected are: W3, F11, E23, T33, M42, T45, Y65, D68, R72, R84, K85, F89, and V93. A better overview of the distribution of TEMPOL attenuations is obtained by plotting these effects on the surface of the protein. Figure 3 ▶ shows two views (Fig. 3A,B) of the surface contact plot, calculated by MOLMOL (Koradi et al. 1996), of the structure of MNEI showing the location of residues whose α–β correlations are most affected by TEMPOL. All atoms of residues with attenuations (2.0< Ai <1.8) are painted in violet, those of residues with attenuations (1.8< Ai <1.4) are painted in pink. Residues whose attenuation is below average are represented in cyan.
Fig. 3.

Surface contact plot of the structure of MNEI showing the location of residues whose α–β correlations are most affected by TEMPOL in the TOCSY spectrum (A,B) or in the ePHOGSY–NOE–TOCSY spectrum (C) and (D). Color code: Violet for residues with attenuations (2.0< Ai< 1.8), pink for residues with attenuations (1.8< attenuation <1.4), cyan for residues with attenuations <0.4; blue and red for ePHOGSY–NOE–TOCSY signals, respectively, with high and low TEMPOL attenuations. Surfaces (E) and (F) show residues (colored in yellow) whose mutation has a major effect on sweetness.
The shape of the protein is approximately that of a discoid: The two views shown in Figure 3 ▶ correspond to the largest areas of the discoid. TEMPOL accessible sites are present on both sides and involve, in addition, two residues on the right-hand border, Y65 and D68. These residues flank the region corresponding to TyrA13-AspA16 of native monellin (i.e., the putative sweet finger previously identified in ELISA tests [Kim et al. 1991]). An outstanding feature of the distribution of accessible areas is the difference among the three main loops of the protein. If one adopts the nomenclature of Murzin (1993) for the cystatin superfamily the loops can be named L23, L34, and L45. In spite of the fact that L23 and L45 are completely exposed (i.e., they have very few interactions with other parts of the protein), they appear less accessible to TEMPOL than L34, the loop corresponding to the mentioned sweet finger.
Thus, for MNEI, such as in the cases of lysozyme (Esposito et al. 1992), bovine pancreatic trypsin inhibitor (BPTI) (Molinari et al. 1997), and tendamistat (Scarselli et al. 1999) surface mapping by TEMPOL-attenuated-2D spectroscopy seems to yield results consistent with biological data. However, before drawing conclusions, it must be kept in mind that this approach may reveal only part of a wide range of surface properties.
Measurement of NOEs between protein and bound water
As mentioned in the introduction, accessibility of the protein surface can ultimately be linked to competitive binding of various ligands with water molecules. The influence of hydration water on the surface properties of proteins has been studied both by solid state and solution methods. The picture obtained from X-ray studies, although static, is a good reference for subsequent NMR-based surface mapping. A recent low-temperature (100°K) study of β-Trypsin, designed to minimize the influence of crystal packing (Nakasako 1999), has shown that >80% of the surface is covered by large-scale networks of water molecules. The same study has also shown that the active site, rather than binding its own water molecules, tends to be exposed to bulk water.
Solution methods that can reveal the presence of bound water molecules can complement TEMPOL accessibility very effectively. The possibility of direct measurement of NOEs between bound water and protein residues was first demonstrated by the pioneering work of Otting and Wüthrich (1989). Acquisition of these experiments is hampered by radiation damping and demagnetizing field mechanisms (Otting 1997), but it is possible to circumvent these problems by using pulsed field gradients (Dalvit and Hommel 1995; Dalvit 1996, 1998; Melacini et al. 1999a, b). For instance, it has been recently shown that NOEs between protons of protein residues and bound water can be measured by a series of H2O-selective experiments called ePHOGSY (Dalvit 1996, 1998) that allow rapid detection of water bound to proteins. The pulsed field gradients in these experiments are used only to destroy unwanted magnetization and for water suppression. 1D ePHOGSY, with NOE or ROE steps, are ideally suited to discriminate between Overhauser and exchange effects. Homonuclear 2D ePHOGSY-NOE-TOCSY spectra were used for a quantitative evaluation of distances between protein and bound water protons.
Intensities of the ePHOGSY correlations depend on the distance between water and protein protons, on protein dynamics, and on the residence time of the water on the protein. Accordingly, it might be possible, in principle, to describe static and dynamic aspects of surface hydration. In practice, it turns out that it is difficult to factorize the different effects, but it is fair to state that, provided exchange phenomena are not prevalent, ePHOGSY correlations reveal the presence, at protein sites, of firmly bound water. In addition, if we perform an ePHOGSY experiment in the presence of TEMPOL, we see attenuations caused by either TEMPOL accessible water molecules while they are bound to a residue or to TEMPOL accessible residues while they are bound to water. Thus ePHOGSY-coupled-TEMPOL-attenuated-1D and 2D spectroscopy is a novel NMR method that allows accurate detection of protein accessibility simultaneously to two different probes (i.e., water and TEMPOL molecules). Figure 4 ▶ shows the 2D ePHOGSY–NOE–TOCSY spectra of MNEI, recorded in diamagnetic (Fig. 4A ▶) and paramagnetic (Fig. 4B ▶) water solutions, and the different extent of the paramagnetic attenuations is here apparent.
Fig 4.

Comparison of the 2D ePHOGSY–NOE–TOCSY spectra of MNEI, recorded in diamagnetic (A) and paramagnetic (B) water solutions. NMR spectra were recorded on 2.0 mM samples in an H2O/2H2O (90:10, v.v) solution containing 18.5 mM potassium phosphate, adjusted to pH 2.9, at 35°C. The paramagnetic samples are also 16 mM in TEMPOL. Arrows and labels refer only to signals that are unambiguously assigned.
There are a total of 43 correlations, including four signals originating from exchange phenomena, as suggested by the positive 1D ePHOGSY–ROE correlations of Y58Hδ1, Y63Hδ1, W3Hɛ3, and K17NH protons. It is important to note that peaks involving serines and threonines and tyrosines may be partly cause by relayed exchange via the OH groups (Otting 1997).
An overview of TEMPOL attenuated ePHOGSY-NOE-TOCSY correlations can be best appreciated by plotting them on the protein surface. Figure 3 ▶ shows two views (Fig. 3C,D ▶) of the surface of MNEI in which residues close to bound water molecules and attenuated by TEMPOL are painted in blue or red according to their relative paramagnetic attenuation.
The area covered by TEMPOL attenuated ePHOGSY–NOE–TOCSY correlations is larger than that covered by TEMPOL attenuated TOCSY correlations, but there is little overlap between affected residues. It can be seen that only two `hot spots' include ePHOGSY–NOE–TOCSY correlations, in the case of T33 and T45 residues, whereas all the others are flanked by them.
Discussion
Several general features of the surface of MNEI can be grasped at first sight from an overall analysis of the NMR data. There are regions, well exposed to solvent, that are silent both with respect to TEMPOL alone or to the combination of ePHOGSY/TEMPOL. The clearest example is the case of the nonnative loop (centered around G51-F52), as suggested by the very low paramagnetic effect experienced by F52 protons. Then there are accessible regions that are revealed only by TEMPOL (e.g., the 65–68 finger contained in the L34 loop). Furthermore, several accessible regions are revealed only by highly attenuated ePHOGSY-NOE-TOCSY signals, as in the case of residues I8, N14, K25, K43, T81, K85, and R88. Surface accessibility is revealed both by ePHOGSY and TEMPOL, only in the case of residues T33 and T45.
The highest TEMPOL induced attenuations of the α–β correlations in the TOCSY spectrum (i.e., those with Ai values higher >1.8) reveal the protein sites where there is a low competition of water molecules with the paramagnetic probe to approach the surface. This interpretation, previously proposed for tendamistat (Scarselli et al. 1999), is here experimentally proven. All the α and β protons of the most TEMPOL accessible residues (i.e., T33, Y65, D68, R84, K85, and V93) are paralleled by very few water-protein NOEs in the ePHOGSY spectra. In fact, observable signals are present only for T33Hα and K85Hβ protons.
This finding cannot be simply explained by a lack of water molecules close to the latter residues because all of them, with the exception of V93, are very hydrophilic. Then, the absence of sizeable water–protein NOEs has to be attributed to dynamic aspect of the water–protein interaction.
The observed intermolecular NOEs in the ePHOGSY–NOE spectra are all negative (i.e., positive signals). This feature implies that, at a Larmor frequency of 600 MHz, the residence time of these water molecules must be >300 ps (Halle et al. 1999). This is certainly the case of water molecules firmly bound to a protein, such as MNEI, which has a τC of the order of 5.1 ns (Spadaccini et al. 2001). For water molecules on the MNEI surface, the effective correlation time would approach 10 s–12 s (i.e., values typical of bulk water). This would yield indeed weak positive NOEs, as in the case of many of the solvent exposed MNEI protons. This behavior, in turn, is consistent with reduced competition of water with TEMPOL.
It should also be discussed that there are protein sites that are not accessible to the spin-label in spite of their exposed topology such as residues R31and R39 (Fig. 2 ▶). The most natural explanation is that they occupy hydration sites where water molecules with long residence times prevent the access of TEMPOL, as shown by the presence of the R31He and R39NH signals in the ePHOGSY–NOE–TOCSY spectrum.
Furthermore, the observation that all signals of the ePHOGSY–NOE–TOCSY spectrum of MNEI experience a sizeable intensity decrease on additions of TEMPOL, albeit with different levels of attenuation, is in agreement with the absence of buried water molecules in the crystal structure of closely related SCM (Somoza et al. 1993). Indeed, should any buried water molecule be present, a large number of signals with very small paramagnetic attenuations should be expected. This is not the case for MNEI.
In Figure 3 ▶, it is possible to see that there are regions where TEMPOL and water molecules approach the protein surface efficiently and to similar extents. From the ePHOGSY–NOE–TOCSY spectra, additional TEMPOL interaction sites can be identified because of the intrinsic good resolution of this experiment. The exclusion of the spin label from a close approach to the MNEI surface is most likely determined by the local stability of the solvation shell.
Thus, complementary information can be obtained from TOCSY and ePHOGSY–NOE–TOCSY spectroscopies in the presence TEMPOL. Three limiting conditions for the protein surface accessibility can be consistently proposed: Regions of spin-label preferential approaches, regions visited both by TEMPOL and water molecules, and regions occupied only by solvent molecules. From a functional point of view, the first case is the most interesting because the active MNEI moiety should be searched among the surface sites with the highest TEMPOL accessibility (Scarselli et al. 1999) and without tightly bound water molecules.
Conclusions
In this study we have attempted to locate the areas of interaction of a sweet protein at its receptor, by means of purely physical methods without prior biological knowledge.
NMR-based techniques, which can highlight solute–solute and solute–solvent interactions, can be regarded as a strategic window to observe the surface characteristics of a protein without the possible artifacts caused by molecular packing in the crystal. In this respect a comparison of water–protein Overhauser effects obtained in the absence and in presence of TEMPOL, as in this report, with the limited number of signals present in the spectrum, easy to be assigned and quantified, is a powerful tool for investigating protein surface features. Reliable information on the protein surface accessibility is obtained by the analysis of the effects of water and nitroxide molecules as different and simultaneous probes.
The main results of our mapping are: Protons of the residues that are most TEMPOL accessible are never tightly bound to water molecules; ePHOGSY/TOCSY spectra indicate that only a limited number of water molecules exhibit residence times on the MNEI surface that are long enough to yield sizeable (negative) NOEs; a combined use of the TEMPOL approach with the ePHOGSY/TOCSY spectroscopy suggests that on the protein surface there are regions of highly ordered water molecules that prevent efficient paramagnetic attenuations of the NOE cross-peaks. It is interesting to note that, among these regions, the largest hydrated patch is on the opposite side with respect to the sweet finger.
Several mutants of SCM and monellin (Somoza et al. 1995) show a substantial loss of activity. The combination of the location of mutated residues with surface accessible areas can aid in the localization of the main interaction points of MNEI with the receptor. The most dramatic mutations are D7/G and G9/A, leading to a loss of three orders of magnitude, and I6/G, R72/G and R88/G leading to a loss of two orders of magnitude. Deletion of the three C-terminal residues decreases sweetness by one order of magnitude. We reported these mutations on the surface of MNEI in Figure 3E ▶, F for an easy comparison of this functional information with the entire set of our data.
All the latter critical residues seem to be very accessible ones, as they do not have tightly bound water with the only exceptions of R72, R88, and P94. However, these residues correspond to sites that are accessible also to TEMPOL, as in the case of R72, one of the most TEMPOL-influenced residues. The paramagnetic probe accessibility and the functional relevance of the hydrated R72, R88, and P94 residues are consistent with the absence of strong hydration sites nearby.
It can be concluded that our study circles out, as mentioned above, the sweet finger located on the tip of loop L34 as the most likely interaction area, but is also consistent with previous mutagenesis studies that pinpointed D7, G9, I6, R72, and R88 as additional interacting residues.
As a general final remark, it should be noted that TEMPOL, because of the reactivity of its N-oxyl moiety (Couet et al. 1985), should not be used to map out the surface accessibility of proteins exhibiting redox activity.
A second aspect that should be taken into account for the proposed TEMPOL-based investigation is the protein concentration. The random nature of the TEMPOL–protein interactions and the long electronic relaxation time of the probe (Schwartz et al. 1979) cause only weak paramagnetic effects in the presence of submillimolar protein concentrations. Therefore, when very diluted protein solutions are needed, alternative paramagnetic probes would be necessary.
Materials and methods
Sample preparation
The recombinant polypeptide of MNEI was expressed in Escherichia coli strain BL21(DE3) cells as described in Spadaccini et al. (2001). After sonication and centrifugation of the cellular pellet suspended in 25 mM sodium acetate, pH 5.5, the protein, fully present in the supernatant, can be purified by ion exchange chromatography on a S-sepharose column with sodium acetate and a sodium chloride concentration gradient. Final purification can be achieved by gel filtration on a G-75 column, using ammonium bicarbonate 150 mM as eluent.
TEMPOL (4-hydroxy-2,2,6,6-tetramethyl-piperidine-1-oxyl) obtained from Sigma was used without any further manipulation.
NMR spectroscopy
Spectra of MNEI alone or in the presence of suitable amounts of TEMPOL were recorded in solution conditions (pH 2.9, 35°C) identical to those employed in our previous study of MNEI (Spadaccini et al. 2001). Unlabeled recombinant protein (Tancredi et al. 1992) was dissolved in the appropriate volume of an H2O/2H2O (90:10, v.v.) solution containing 18.5 mM potassium phosphate, to make a 2.0 mM sample, adjusted to pH 2.9, at 35°C. The paramagnetic NMR samples contained 2 mM monellin and an optimal TEMPOL concentration, which was achieved at 16 mM. This condition was reached by adding directly to the NMR tube few microliters of a 2 M TEMPOL solution.
NMR measurements, run at 35°C to reproduce the experimental conditions of the original structural study (Spadaccini et al. 2001), were performed on a Bruker DRX600 spectrometer. Data processing was performed with NMRPipe (Delaglio et al. 1995) and spectral analysis with NMRView (Johnson and Blevins 1994). The 1H chemical shifts are relative to trimethylsilylpropionic 2,2,3,3-d4 acid sodium salt (TSP) as 0.0 ppm.
Resonance assignments were obtained from 2D experiments. A conventional set of 2D spectra, according to the scheme of sequential assignment described by Wüthrich (1986) was recorded: COSY (Aue et al. 1976), TOCSY (Bax and Davis 1985), and NOESY (Jeener et al. 1979). Standard pulse sequences were used to obtain TOCSY spectra (Rance 1987) with mixing times of 54 ms. Water resonance was attenuated using a DANTE presaturation train superimposed to the specific sequence. A total of 470 increments were collected in t1 with 1908 data points and 64 scans/FID in t2, over a spectral width of 6 kHz in both dimensions. Prior to 2D FT the experimental array was zero-filled to a final matrix of 2048×1024 data points. 1D ePHOGSY with NOE and with ROE, to discriminate between Overhauser and exchange effects and 2D ePHOGSY-NOE-TOCSY (Dalvit. 1996, 1998), for a quantitative analysis of the effects, were generated. In both 1D PHOGSY experiments, spectra were acquired with 16384 data points and 4096 scans, whereas the 2D ePHOGSY–NOE–TOCSY were obtained with 256 increments and 320 scans over 2048 data points. After zero filling, the final matrix was 4096 × 512 data points. In all the ePHOGSY experiments, the mixing time to build up the intermolecular Overhauser effects was 202 ms. The H2O-selective 180° Gaussian pulse between the first two pulsed field gradients in all the ePHOGSY sequences was achieved according to Dalvit (1996) with a duration of 50 ms and 60 dB of attenuation. Water suppression was achieved following the scheme of Hwang and Shaka (1995).
Attenuations
As previously reported (Molinari et al. 1997), paramagnetic effects were measured by comparing autoscaled cross-peak attenuation figures, Ai, defined as:
 |
1 |
That is to say that the individual deviations from the average of the cross-peak autoscaled volumes, Vip, d, the latter defined as:
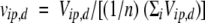 |
2 |
where n is the number of measured cross-peak volumes and Vid and Vip are the protein individual cross-peak volumes measured in the absence and in the presence of the spin probe, respectively.
The individual Ais are plotted versus protein-sequence position and the values lying above or below the average attenuation level, unitary by construction because (Σivip,d/n) = 1, correspond respectively to high or low spin-probe accessibility levels (Molinari et al. 1997). With this representation it is easy to compare experiments performed in different conditions (temperature, protein, and paramagnetic probe concentration, solvents) because any general effect is included in the mean value and the observed deviations. Because of the experimental errors in measuring the cross-peak volumes, the standard deviation analysis defines the range of confidence of the attenuation data. Therefore, only the paramagnetic attenuation values outside the standard deviation are worth considering. For nonequivalent methylene protons a mean attenuation value was reported. Ai values have been compared to the sum of the autoscaled exposed surface areas (ESAi; Molinari et al. 1997). The exposed surface area, ESAi, calculated with MOLMOL (Koradi et al. 1996), in analogy to the Ai definition was used to obtain the corresponding autoscaled value.
For each methyl and methylene group the sum of the individual hydrogen exposures was considered.
All displays of structures, were carried out with the program MOLMOL (Koradi et al. 1996).
Acknowledgments
We thank the Italian Ministry of Research and Technology (PRIN 1997 and 1999) and the University of Siena for financial support. P.A.T. wishes to thank Da-guin for inspiring suggestions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ELISA, enzyme linked assay
ePHOGSY, enhanced protein hydration observed through gradient spectroscopy
NMR, nuclear magnetic resonance
NOE, nuclear Overhauser effect
NOESY
NOE spectroscopy
SAR, structure-activity relationship
TEMPOL,4-hydroxy-2,2,6,6-tetramethyl-piperidine-1-oxyl
TOCSY, total correlation spectroscopy
ppm, parts per million
r.m.s.d., root-mean-square deviation
1D, monodimensional
2D, two dimensional
3D, three dimensional
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.30101
References
- Ariyoshi Y. and Kohmura, M. 1994. Solid-phase synthesis and structure-activity relationships of analogs of the sweet protein monellin. J. Soc. Synth. Org. Chem. Jpn. 52 359–369. [Google Scholar]
- Aue, W.P., Bartholdi E., and Ernst, R.R. 1976. Two dimensional spectroscopy. Application to nuclear magnetic resonance. J. Chem. Phys. 64 2229–2246. [Google Scholar]
- Bax, A. and Davis, D.G. 1985. Mlev-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65 355–360. [Google Scholar]
- Caldwell, J.E., Abildgaard, F., Dzakula, Z., Ming, D., Hellekant, G., and Markley, J.L. 1998. Solution structure of the thermostable sweet-tasting protein brazzein. Nat. Struct. Biol. 5 427–431. [DOI] [PubMed] [Google Scholar]
- Couet, W.R., Brasch, R.C., Sosnovsky, G., Lukszo, J., Prakash, I., Gnewuch, C.T., and Tozer, T.N. 1985. Influence of chemical structure of nitroxyl spin labels on their reduction by ascorbic acid. Tetrahedron 41 1165–1172. [Google Scholar]
- Dalvit, C. 1996. Homonuclear 1D and 2D NMR experiments for the observation of solvent-solute interactions. J. Magn. Reson. B. 112 282–288 [DOI] [PubMed] [Google Scholar]
- Dalvit, C. 1998. Efficient multiple-solvent suppression for the study of the interaction of organic solvents with biomolecules. J. Biomol. NMR 11 437–444. [Google Scholar]
- Dalvit, C., Floersheim, P., Zurini, M., and Widmer, A. 1999. Use of organic solvents and small molecules for locating binding sites on proteins in solution. J. Biomol. NMR 14 23–32 [DOI] [PubMed] [Google Scholar]
- Dalvit, C. and Hommel, U. 1995. Sensitivity-improved detection of protein hydration and its extension to the assignment of fast-exchanging resonances. J. Magn. Reson. 109 334–338 [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensioal spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Esposito, G., Lesk, A.M., Molinari, H., Motta, A., Niccolai, N., and Pastore, A. 1992. Probing protein structure by solvent perturbation of nuclear magnetic resonance spectra. Nuclear magnetic resonance spectral editing and topological mapping in proteins by paramagnetic relaxation filtering. J. Mol. Biol. 224 659–670. [DOI] [PubMed] [Google Scholar]
- Goodman, M., Coddington, J., and Mierke, D.F. 1987. A model for the sweet taste of stereoisomeric retro-inverso and dipeptide amides. J. Amer. Chem. Soc. 109 4712–4714. [Google Scholar]
- Halle, B., Denisov, V.P., and Venu, K. 1999. Multinuclear relaxation dispersion studies of protein hydration in biological magnetic resonance, volume 17 419–484: Structure computation and dynamics in protein NMR (eds. N.R. Krishna and L.J. Berliner) Kluwer Academic/Plenum Publishers, New York.
- Harada, S., Otani, H., Maeda, S., Kai, Y., Kasai, N., and Kurihara, Y. 1994. Crystallization and preliminary X-ray diffraction studies of curculin. A new type of sweet protein having taste-modifying action. J. Mol. Biol. 238 286–287. [DOI] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1995. Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 20 478–480. [DOI] [PubMed] [Google Scholar]
- Hung, L.W., Kohmura, M., Ariyoshi, Y., and Kim, S.H. 1999. Structural differences in D and L-monellin in the crystals of racemic mixture. J. Mol. Biol. 8 311–321. [DOI] [PubMed] [Google Scholar]
- Hwang, T.L. and Shaka, A.J. 1995. Water suppression that works. Excitation sculpting using arbitrary waveforms and pulsed field gradients. J. Magn. Reson. A112 275–279. [Google Scholar]
- Inglett, G.E. and May, J.F. 1969. Serendipity berries source of a new intense sweetener. J. Food Sci. 34 408–411. [Google Scholar]
- Iwamura, H. 1981. Structure-sweetness relationship of L-aspartyl dipeptide analogues. A receptor site topology. J. Med. Chem. 24 572–578. [DOI] [PubMed] [Google Scholar]
- Jeener, J., Meyer, B.H., Bachman, P., and Ernst, R.R. 1979. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 71 4546–4553. [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMR View: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4 603–614. [DOI] [PubMed] [Google Scholar]
- Kier, L.B. 1972. Molecular theory of sweet taste. J. Pharm. Sci. 61 1394–1397. [DOI] [PubMed] [Google Scholar]
- Kim, S.H., Kang, C.H., and Cho, J.M. 1991. Sweet proteins: biochemical studies and genetic engineering. In Sweeteners, discovery, molecular design and chemoreception. (eds., D.E. Walters, F.T. Orthofer, and G.E. DuBois) pp. 28–40. ACS Symposium Series 450, ACS, Washington, DC.
- Kim, S.H., Kang, C.H., Kim, R., Cho, J.M., Lee, Y.B., and Lee, T.K. 1989. Redesigning a sweet protein: increased stability and renaturability. Protein Engineering 2 571–575. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structure. J. Mol. Graph. 14 51–55. [DOI] [PubMed] [Google Scholar]
- Krantz, A. 1998. Probing protein surfaces for `hot spots': A new frontier. TIBTECH 16 198–199. [Google Scholar]
- Kurihara, K. and Beidler, L.M. 1968. Taste-modifying protein from miracle fruit. Science 161 1241–1243. [DOI] [PubMed] [Google Scholar]
- Liepinsh, E. and Otting, G. 1997. Organic solvents identify specific ligand binding sites on protein surfaces. Nature Biotech. 15 264–268. [DOI] [PubMed] [Google Scholar]
- Liu, X., Maeda, S., Hu, Z., Aiuchi, T., Nakaya, K., and Kurihara, Y. 1993. Purification, complete amino acid sequence and structural characterization of the heat-stable sweet protein, mabinlin II. Eur. J. Biochem. 211 281–287. [DOI] [PubMed] [Google Scholar]
- Mattos, C. and Ringe, D. 1996. Locating and characterizing binding sites on proteins. Nature Biotech. 14 595–599. [DOI] [PubMed] [Google Scholar]
- Melacini, G., Boelens, R., and Kaptein, R. 1999a. Band-selective editing of exchange-relay in protein-water NOE experiments. J. Biomol. NMR 13 67–71. [DOI] [PubMed] [Google Scholar]
- Melacini, G., Boelens, R., and Kaptein, R. 1999b. Water-macromolecule interactions by NMR: A quadrature-free constant-time approach and its application to CI2. J. Biomol. NMR 15 189–201. [DOI] [PubMed] [Google Scholar]
- Ming, D. and Hellekant, G. 1994. Brazzein, a new high-potency thermostable sweet protein from Pentadiplandra brazzeana B. FEBS Lett. 355 106–108. [DOI] [PubMed] [Google Scholar]
- Molinari, H., Esposito, G., Pegna, M., Ragona, L., Niccolai, N., and Zetta, L.. 1997. Probing protein structure by solvent perturbation of NMR spectra: The surface accessibility of bovine pancreatic trypsin inhibitor. Biophys. J. 73 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncrieff, R.W. 1967. The chemical senses. Hill: London.
- Morris, J.A. 1976. Sweetening agents from natural sources. Lloydia 39 25–38. [PubMed] [Google Scholar]
- Murzin, A.G. 1993. Sweet-tasting protein monellin is related to the cystatin family of thiol proteinase inhibitors. J. Mol. Biol. 230 689–694. [DOI] [PubMed] [Google Scholar]
- Nakasako, M. 1999. Large-scale networks of hydration water molecules around bovine β-trypsin revealed by cryogenic X-ray crystal structure analysis. J. Mol. Biol. 289 547–564. [DOI] [PubMed] [Google Scholar]
- Otting, G. 1997. NMR studies of water bound to biological molecules. Prog. NMR Spectrosc. 31 259–285. [Google Scholar]
- Otting, G. and Wüthrich, K. 1989. Studies of protein hydration in aqueous solution by direct NMR observation of individual protein-bound water molecules. J. Am. Chem. Soc. 111 1871–1875. [Google Scholar]
- Rance, M. 1987. Improved technique for homonuclear rotating frame and isotropic mixing experiments. J. Magn. Reson. 74 557–564. [Google Scholar]
- Ringe, D. 1995. What makes a binding site a binding site? Curr. Opin. Struct. Biol. 5 825–829. [DOI] [PubMed] [Google Scholar]
- Savvides, S.N. and Karplus, P.A. 1996. Kinetics and crystallographic analysis of human glutathione reductase in complex with a xanthene inhibitor. J. Biol. Chem. 271 8101–8107. [DOI] [PubMed] [Google Scholar]
- Scarselli, M., Bernini, A., Segoni, C., Molinari, H., Esposito, G., Lesk, A.M., Laschi, F., Temussi, P.A., and Niccolai, N. 1999. Tendamistat surface accessibility to the TEMPOL paramagnetic probe. J. Biomol. NMR 15 125–133. [DOI] [PubMed] [Google Scholar]
- Schwartz, R.N., Jones, L.L., and Bowman, M.K. 1979. Electron spin-echo studies of nitroxide free radicals in liquids. J. Phys. Chem. 83 3429–3434. [Google Scholar]
- Shallenberger, R.S. and Acree, T. 1967. Molecular theory of sweet taste. Nature 216 480–482. [DOI] [PubMed] [Google Scholar]
- Somoza, J.R., Cho, J.M., and Kim, S.H. 1995. The taste-active regions of monellin, a potently sweet protein. Chem. Senses 20 61–68. [DOI] [PubMed] [Google Scholar]
- Somoza, J.R., Jiang, F., Tong, L., Kang, C.H., Cho, J.M., and Kim, S.H. 1993. Two crystal structures of a potently sweet protein. Natural monellin at 2.75 Å resolution and single-chain monellin at 1.7 Å resolution. J. Mol. Biol. 234 390–404. [DOI] [PubMed] [Google Scholar]
- Spadaccini, R., Crescenzi, O., Tancredi, T., De Casamassimi, N., Saviano, G., Scognamiglio, R., Di Donato, A., and Temussi, P.A. 2001. Solution structure of a sweet protein: NMR study of MNEI, a single chain monellin. J. Mol. Biol. 305 505–514. [DOI] [PubMed] [Google Scholar]
- Tancredi, T., Iijima, H., Saviano, G., Amodeo, P., and Temussi, P.A. 1992. Structural determination of the active site of a sweet protein: A 1H NMR investigation of MNEI. FEBS Lett. 310 27–30. [DOI] [PubMed] [Google Scholar]
- Temussi, P.A., Lelj, F., and Tancredi T. 1991. Structure-activity relationship of sweet molecules. In Sweeteners, discovery, molecular design and chemoreception. (eds., D.E, Walters, F.T. Orthofer, G.E. DuBois), pp. 143–161. ACS Symposium Series 450, ACS, Washington, DC..
- Temussi, P.A., Lelj, F., and Tancredi, T. 1978. Three-dimensional mapping of the sweet taste receptor site. J. Med. Chem. 21 1154–1158. [DOI] [PubMed] [Google Scholar]
- Temussi, P.A., Lelj, F., Tancredi, T., Castiglione-Morelli, M.A., and Pastore, A. 1984. Soft agonist-receptor interactions: Theoretical and experimental simulation of the active site of the receptor site of sweet molecules. Int. J. Quantum Chem. 26 889–906. [Google Scholar]
- Van der Wel, H. and Loeve, K. 1972. Isolation and characterisation of thaumatin I and II, the sweet-tasting proteins from Thaumatococcus daniellii. Eur. J. Biochem. 31 221–225. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids. Wiley, New York.
- Zaffaroni, A. 1975. U.S. Patent 3 876 816, CA.