

Abstract
This study shows that a combination of sequence homology and structural information can be used to increase the stability of the WW domain by 2.5 kcal mol−1 and increase the Tm by 28°C. Previous homology-based protein design efforts typically investigate positions with low sequence identity, whereas this study focuses on semi-conserved core residues and proximal residues, exploring their role(s) in mediating stabilizing interactions on the basis of structural considerations. The A20R and L30Y mutations allow increased hydrophobic interactions because of complimentary surfaces and an electrostatic interaction with a third residue adjacent to the ligand-binding hydrophobic cluster, increasing stability significantly beyond what additivity would predict for the single mutations. The D34T mutation situated in a π-turn possibly disengages Asn31, allowing it to make up to three hydrogen bonds with the backbone in strand 1 and loop 2. The synergistic mutations A20R/L30Y in combination with the remotely located mutation D34T add together to create a hYap WW domain that is significantly more stable than any of the protein structures on which the design was based (Pin and FBP28 WW domains).
Keywords: WW domain, protein design, homology-based, sequence alignment, structure comparison, protein stability, side-chain interactions
Protein design focuses on generating sequences that can adopt a unique ensemble of closely related three-dimensional structures. Many advances have been made in this field, including the early design of simple protein secondary structures by using molecular modeling, and more recently the use of computational algorithms to design proteins (DeGrado et al. 1989; Hecht et al. 1990; Street and Mayo 1999; Baker 2000). The success of any rational design strategy largely depends on the energy force fields on which it is based. Most force fields consider van der Waals forces, the hydrophobic effect, electrostatic interactions, and conformational entropy (Shakhnovich and Gutin 1993; Desjarlais and Handel 1995; Dahiyat et al. 1997; Koehl and Levitt 1999; Jiang et al. 2000). Although our understanding of the forces stabilizing protein structures continues to evolve, computational protein design still faces limitations imposed by the requirement for a high-resolution crystal structure as the starting point. Even though there are >15,000 structures available, only 1500 are crystal structures with a resolution below 1.7 Å, suitable for structure-based design. On the other hand, rapidly emerging protein sequence information (>500,000 sequences) as well as structural information accumulating in the databases offer the opportunity to look at proteins as an entire family. Studies of several protein families have shown significant sequence and structural conservation in the hydrophobic core and in positions that are involved in binding or are otherwise functionally important (Bashford et al. 1987; Lesk and Fordham 1996; Chothia et al. 1998; Michnick and Shakhnovich 1998; Larson and Davidson 2000). In this paper, we use such information for the design of stable proteins. We take advantage of the wealth of information about native sequences in design (so-called homology-based protein design [Nikolova et al. 1998; Wang et al. 1999; Perl et al. 2000]), in combination with traditional structure-based design that uses structural alignments (Rufino and Blundell 1994).
We chose the WW domain to test this design approach because of our keen interest in understanding the forces that stabilize β-sheet structure. This domain represents the shortest sequence (38–40 amino acids) that forms a monomeric three-stranded single-layer β-sheet that folds and unfolds cooperatively without cofactors or disulfide bonds (Sudol 1996; Koepf et al. 1999b; Crane et al. 2000). It appears in >170 proteins and mediates protein–protein interactions by interacting with proline-containing ligands (Sudol and Hunter 2000). Two high-resolution crystal structures, including a 1.35-Å structure of the peptidyl-prolyl cis-trans isomerase (Pin) WW domain (Ranganathan et al. 1997; Huang et al. 2000), and several NMR structures, including the human Yes-associated protein WW domain (hYap) bound to a proline-rich ligand (Macias et al. 1996), as well as the formin-binding protein (FBP28) WW domain (Macias et al. 2000) are available from the Protein Data Bank. WW domains contain two strictly conserved tryptophan residues spaced 20–22 amino acids apart in the sequence for which they are named, as well as a highly conserved C-terminal proline residue. Previous studies on the hYap WW domain show that the N-terminal tryptophan (Trp17) is critical for the folding of the protein, whereas the C-terminal tryptophan (Trp39) is essential for ligand binding (Macias et al. 1996; Koepf et al. 1999a). Despite the similarities in the tertiary structures of the WW domains, both experimental studies and molecular dynamics simulations show that the thermodynamic stabilities of these small proteins can differ dramatically (Ibragimova and Wade 1999; Macias et al. 2000). For example, the hYap WW domain (57 residues) has a Tm of 51°C, whereas FBP28 (37 residues) and Pin (34 residues) exhibit higher thermal stabilities with Tm's of 62° and 58°C, respectively (M. Jäger and J.W. Kelly, unpubl.). Several shorter constructs of the hYap WW domain are either less stable than the 57-mer or are unfolded (E. Koepf and J.W. Kelly, unpubl.). It was reported that the side chain of an isoleucine residue at position 7 covers a hydrophobic patch in the hYap WW domain and is important for its stability (Macias et al. 1996).
Structural information from WW domain proteins identifies two segregated hydrophobic clusters, one on each side of the three-stranded β-sheet. The first cluster of the hYap WW domain is composed of the side chains of the N-terminal Trp (W17) as well as those of Pro14, Phe29, Asn31, Thr38, and Pro42. Cluster 2 resides on the ligand-binding face and is more hydrophobic, involving the side chains of residues Glu18, Ala20, Tyr28, Leu30, His32, Thr37, and Trp39. Most of the residues in these two clusters are semi-conserved among WW domains. Figure 1 ▶ lists the sequences of the WW domains from hYap, Pin, and FBP28, as well as the consensus sequence generated using the SMART server (http://smart.embl-heidelberg.de) (Schultz et al. 1998, 2000) based on 166 globular proteins, containing >200 nonredundant WW domain sequences.
Fig. 1.

Sequence of the hYap, Pin, and FBP28 WW domains (top) as well as the consensus sequence generated by the SMART server (http:// smart.embl-heidelberg.de) (bottom). Wave-underlined residues compose hydrophobic cluster 1 in the hYap sequence, whereas doubly underlined residues belong to hydrophobic cluster 2 (see text). Boldface letters identify the residues at positions 20, 30, and 34 in the hYap WW domain, which are mutated to increase the protein stability. In the consensus sequences, the single-letter amino acid codes are in capital letters; lowercase letters are defined as follows: (h) hydrophobic; (a) aromatic; (p) polar; (o) alcohol (Ser or Thr); (t) turn-like residues (Ala, Cys, Asp, Glu, Gly, His, Lys, Asn, Gln, Arg, Ser, and Thr). Positions with low or no sequence conservation are marked with a dot. The definitions were slightly modified from the original output of SMART program to improve readability. Residues involved in either of the hydrophobic clusters that show ≥50% consensus but are not highly conserved are defined as semi-conserved. This study focuses on these semi-conserved core residues and their surrounding residues.
Aside from thermostability differences, WW domains also exhibit different binding selectivities and can be divided into four or five subclasses, depending on the type of proline-rich ligands that they bind (Kay et al. 2000; Verdecia et al. 2000). Subclass I WW domains, including hYap, bind ligands with a PPXY motif. FBP28 belongs to subclass II, which recognizes a PPLP motif (Bedford et al. 1997; Espanel and Sudol 1999). Pin WW domain prefers a (phosphorylated-S/T)P sequence motif and belongs to subclass IV (Sudol and Hunter 2000).
The small size of the WW domain, combined with available sequence and structural information, makes it an ideal model for studying sequence, structure, and function relationships as they relate to a three-stranded β-sheet structure. Our goal was to reengineer the relatively unstable hYap WW domain to afford a more thermostable β-sheet by introducing point mutations based on analysis of both sequence and structure of WW homologs. Mutations were introduced to utilize stabilizing forces such as solvation energy, hydrogen bonding, electrostatic interactions, and van der Waals interactions. Both structural and sequence homologies were considered in the redesign of semi-conserved residues in the two hydrophobic clusters and their surrounding residues.
Results
Design rationale
The mutation D34T enables hydrogen bonding between Asn31 and the protein backbone
The amino acids making up the two hydrophobic clusters are semi-conserved among all WW domain proteins (Fig. 1 ▶), that is, they show ≥50% consensus but are not highly conserved. Because the WW domain is composed of a single-layer β-sheet, most of the core residues are partially exposed. In the first cluster, Trp17 dominates the hydrophobic interactions, as demonstrated from earlier work on hYap (Koepf et al. 1999b). An examination of the peripheral region of this cluster employing the 1.35-Å resolution crystal structure of the Pin WW domain suggests that the side chain of Asn26 plays an essential stabilizing role by forming three hydrogen bonds to the protein backbone (Fig. 2B ▶). Asn26 of the Pin WW domain is a buried polar residue. Solvent-excluded polar residues are in many cases involved in hydrogen bonding and contribute to protein stability (Lumb and Kim 1995; Maxwell and Davidson 1998; Zhu et al. 2000; Pace 2001). The side-chain amide proton of Asn26 hydrogen bonds to the protein backbone via the carbonyl oxygen of Pro9, whereas the Asn26 side-chain amide oxygen forms hydrogen bonds to the backbone amide protons from Ile28 and Thr29. Residues within 4.5 Å of Asn26 (side-chain heavy atoms) are Trp11, Ile28, Thr29, and Ala31. A similar group of contacting residues (Trp17, Ile33, Asp34, and Thr36) are seen around Asn31 (which corresponds to Asn26 in the Pin WW domain) in the NMR structure of the hYap WW domain, with the exception of a change of Asp34 in hYap in the place of a Thr29 in Pin (Fig. 2A ▶). A structural examination, albeit a low-resolution NMR structure, clearly suggests that the side chain of Asn31 in hYap is rotated along the Cβ–Cγ bond relative to Asn26 in the Pin WW domain (Asn31 of hYap has a χ2 angle of −9.4° compared with χ2 of −64.7° for Asn26 in Pin WW domain). This is possibly the result of the strong dipole attraction between the side-chain NH of Asn31 and the negatively charged carboxylate group on the Asp34 side chain. As a result, the Asn31 side-chain amide NH in the wild-type hYap WW domain points toward the side chain of Asp34, interfering with hydrogen-bonding interactions between the Asn31 side chain and the protein backbone (Fig. 2A ▶). The importance of Asn31 in hYap was tested by a N31A mutation, which resulted in a mostly unfolded protein structure according to far-UV circular dichroism (CD) analysis (Fig. 3 ▶).
Fig. 2.

Structural comparison of the environment around the identically located Asn31 in hYap (A) and Asn26 in Pin (B). The numbers on dotted lines indicate non-bonded distances in Å between the two heavy atoms. These asparagine residues adopted different χ2 angles, resulting in different hydrogen bonding patterns around them (see text for more detail).
Fig. 3.

Far-UV CD spectra of the wild-type hYap WW domain (50 μM) at 2°C (·) and at 98°C (+), as well as that of the A20R/L30Y/D34T (▪) and N31A (Δ) WW domains at 2°C. The N31A variant is mostly unfolded, implying the importance of Asn31 for structural integrity at pH 7.
If the hypothesis of the dipole attraction between the Asn31 and Asp34 side chains is correct, a D34T mutation should allow two to three hydrogen bonds to form between the Asn31 side-chain amide group and the protein backbone and consequently increase protein stability, as in the Pin WW domain (Fig. 2B ▶). Interestingly, threonine is the most prevalent type of amino acid at this position, occurring in >50% of the WW domain proteins.
The mutation A20R increases electrostatic and hydrophobic interactions in the second hydrophobic cluster
In the Pin WW domain crystal structure, Arg14 (which is equivalent to Ala20 in hYap; Fig. 4A ▶) forms a salt bridge with Glu12 (which corresponds to a Glu18 in the hYap sequence), which further forms a hydrogen bond with His27 (which corresponds to His32 in hYap; Fig. 4B ▶). Such a hydrogen-bonded network motif is conserved over a large number of proteins. Introduction of Arg at position 20 in hYap should result in formation of a salt bridge with Glu18, with the latter interacting with His32 as well. Therefore, Glu18 can act as a "networking" residue connecting amino acid side chains from different strands in the β-sheet structure. In addition, the n-propyl side-chain substructure of Arg20 buries more hydrophobic solvent-accessible surface area (ΔASAap) against Leu30 (in comparison to Ala20; Fig. 4A ▶), which should increase ΔGu, on the basis of the correlation between increased positive ΔASAap and increased ΔGu (Murphy and Freire 1992; Jiang et al. 2000). Structural comparisons of the second hydrophobic cluster (the ligand-binding site) reveal that the hYap cluster (Fig. 4A ▶) is more exposed than is the cluster in the Pin (Fig. 4B ▶) and FBP28 WW domains, especially at position 30. The buried surface area at this position is 99.0 Å2 for Leu30 in hYap, 141.4 Å2 for Phe25 in Pin, and 138.0 Å2 for the equivalent Tyr22 position in FBP28 (Fig. 1 ▶). The introduction of A20R mutation onto the first strand of hYap should increase solvation energy (hydrophobic interactions) by burying the isobutyl group of Leu30 against the n-propyl substructure in Arg, as suggested by molecular modeling using an optimized set of dihedral angles for the Arg side chain. The amphiphilic side chain of A20R can also bury more surface area against the side chain of Tyr28 in the folded hYap WW domain structure. It is important to point out that the choice of the A20R mutation was based mainly on the consideration of structural homology and not on sequence homologies (Fig. 1 ▶). As a matter of fact, this position was assigned to be Tyr in a designed WW prototype protein, which shows a Tm of 44°C (Macias et al. 2000).
Fig. 4.

Structural comparison of the ligand-binding hydrophobic cluster 2 in hYap (A) and Pin WW (B) domains. The residues composing cluster 2 are depicted by a ball-and-stick representation, with backbone atoms in white, whereas the side-chain atoms are represented with darker shades. The figures were generated using Molscript (Kraulis 1991), on the basis of the NMR structure of hYap (Macias et al. 1996) and the crystal structure of the WW domain from the Pin (Ranganathan et al. 1997, PDB file 1pin).
The hydrophobic mutation L30Y in hydrophobic cluster 2
Leu30 resides in the central strand and is in contact with the Ala20, Tyr28, His32, and Thr37 side chains in the wild-type hYap structure (Fig. 4A ▶). Approximtely 50% of WW domain proteins have aromatic side chains at this position (Fig. 1 ▶). Modeling suggests that the L30Y mutation only modestly increases the buried hydrophobic surface area in the hYap WW domain, because this position is mostly exposed. However, the L30Y increase in buried hydrophobic surface area is more dramatic when made in combination with the A20R mutation discussed above. Modeling shows that a Leu → Tyr mutation at position 30 would complement the increased hydrophobic cavity created by the A20R mutation. The A20R/L30Y double mutation should be synergistic because this arrangement not only greatly increases the buried surface area of the Tyr30 side chain but also reduces the possible conformational space available to the Arg20 side chain, therefore reducing the chain entropy for the arginine side chain. Hence, these mutations should contribute more to stability than additivity would predict.
1H NMR spectroscopy
Well-dispersed chemical shifts were observed in the 1D proton NMR spectra (25°C), both in the amide proton region and in the upfield aliphatic region for all of the designed single- and multiple-mutation-containing WW domains, which is characteristic of structurally well-defined proteins. The amide regions of several representative WW variants are shown in Figure 5 ▶. The indole N-H protons of Trp17 and Trp39 in the wild-type hYap WW domain exhibited chemical shifts of 10.48 and 10.16 ppm, respectively. A downfield chemical shift of the Trp39 indole resonance was observed for the A20R variant, reflective of a more deshielding environment for Trp39, which is not surprising because the mutation is on the same face as the tryptophan in hydrophobic cluster 2. Interestingly, all other variants showed downfield chemical shifts for Trp17 relative to that of wild-type hYap, indicating a more deshielding and perhaps less dynamic hydrophobic core around Trp17 in these variant proteins. This observation suggests that mutations on the opposite side of the β-sheet of Trp17 have a global stabilizing effect.
Fig. 5.

NMR (1D proton) of the amide region of the wild type (A), A20R (B), L30Y (C), D34T (D), and A20R/L30Y/D34T (E). Notice the well-dispersed chemical shifts for the variants. The two dotted lines indicate chemical shifts of the two labeled tryptophan indole NHs in the wild-type hYap WW domain (10.48 and 10.16 ppm).
Thermal stabilities of the mutants
The far-UV CD spectra of all the folded WW variants display a characteristic maximum around 230 nm resulting from the aromatic contribution to the far-UV CD spectrum (Fig. 3 ▶; Koepf et al. 1999a). Upon thermal denaturation of the domains, this peak gradually decreases in intensity until the spectrum resembles a typical random-coil spectra, lacking a maximum at 230 nm (Fig. 3 ▶). The intensity of this peak was therefore used to monitor structural changes during the thermal denaturation transitions (Koepf et al. 1999a,b). All WW variants showed reversible and concentration-independent thermal denaturation/refolding transitions, as expected for monomeric proteins characterized by two-state folding behavior (Fig. 6 ▶). The midpoint of the thermal denaturation curves (Tm) for the three single mutants (D34T, A20R, and L30Y) is 56.5°, 63.0°, and 54.5°C, respectively, higher than the Tm of the wild-type protein (51.6°C). The double mutations (A20R/D34T and A20R/L30Y) and the triple mutation (A20R/L30Y/D34T) significantly increased the Tm to 68.0°, 73.7°, and 79.6°C, respectively (Table 1a,b).
Fig. 6.

Thermal denaturation curves of hYap wild-type and variant WW domains. (A) Molar ellipticity at 230 nm and (B) percentage of unfolding as a function of temperature. The identities of the curves are as follows: (·) wild type; (|ap) L30Y; (×) D34T; (○) A20R; (♦) A20R/D34T; (▴) A20R/L30Y; and (▪) A20R/L30Y/D34T. Percentage of unfolding was calculated according to equation 4 after fitting the original data to equations 2 and 3. Solid lines through the data are the fitting results for (A) and smoothed curves to facilitate viewing for (B).
Table 1.
Thermal stability summary of hYap wild-type and variant WW domainsa (a)
| WW mutants | ΔHm (kcal/mol) | Tm (°C) | ΔTm (°C) | ΔGu (65°C) (kcal/mol) | ΔΔGu (65°C) (kcal/mol) |
| Wild type | 27.0 ± 1.1 | 51.6 ± 0.4 | — | −1.08 | — |
| L30Y | 26.7 ± 0.6 | 54.5 ± 0.2 | 2.9 | −0.91 | 0.17 |
| D34T | 29.2 ± 0.9 | 56.5 ± 0.3 | 4.9 | −0.73 | 0.35 |
| A20R | 29.9 ± 1.4 | 63.0 ± 0.5 | 11.4 | −0.20 | 0.89 |
| A20R/D34T | 34.7 ± 1.0 | 68.0 ± 0.3 | 16.4 | 0.26 | 1.34 |
| A20R/L30Y | 34.5 ± 1.6 | 73.7 ± 00.5 | 22.1 | 0.78 | 1.86 |
| A20R/L30Y/D34T | 40.7 ± 0.5 | 79.6 ± 0.2 | 28.0 | 1.46 | 2.54 |
| WW mutants in 2M NaCl | ΔHm (kcal/mol) | Tm (°C) | ΔTm (°C) | ΔGu (65°C) (kcal/mol) | ΔΔGu (65°C) (kcal/mol) |
| a 50 μM protein in 20 mM potassium phosphate, 100 mM NaCl (pH 7). | |||||
| a 50 μM protein in 20 mM potassium (pH 7). | |||||
| Wild type | 25.0 ± 1.0 | 52.9 ± 0.4 | — | −0.99 | — |
| L30Y | 29.5 ± 0.5 | 60.0 ± 0.4 | 7.1 | −0.45 | 0.54 |
| D34T | 24.7 ± 0.6 | 57.7 ± 0.6 | 4.8 | −0.58 | 0.41 |
| A20R | 20.9 ± 1.2 | 59.3 ± 2.3 | 6.4 | −0.39 | 0.60 |
| A20R/D34T | 29.6 ± 1.1 | 68.4 ± 0.6 | 15.5 | 0.25 | 1.24 |
| A20R/L30Y | 32.6 ± 0.6 | 73.9 ± 0.3 | 21.0 | 0.76 | 1.75 |
| A20R/L30Y/D34T | 34.7 ± 0.7 | 78.2 ± 0.7 | 25.3 | 0.99 | 1.98 |
The unfolding free energies (ΔGu) as well as the differences in free energies (ΔΔGu) were calculated and compared at 65°C (Table 1a), a temperature at which all the variants are in the transition region. Therefore, no extrapolation was needed to derive ΔGu at 65°C, and thus a more accurate comparison can be made among all of the proteins. As expected, the stabilizing contribution of the D34T mutation was additive when introduced with the A20R or the A20R/L30Y mutations, because additivity is often observed when mutations are not in contact with each other (Dill 1997; Wells 1990). Additivity was not expected when both the A20R and L30Y mutations were introduced, because they bury their hydrophobic surfaces into each other in hydrophobic cluster 2. As shown in Table 1a, the double mutation A20R/L30Y contributed more to protein stability than additivity would allow us to predict.
The thermal stability of the mutants was also measured at a high salt concentration (2 M NaCl), and the results are displayed in Table 1b. The stabilizing influence (ΔGu at 65°C) of the A20R mutation under high salt conditions was reduced to ∼65% of the effect in standard buffer (20 mM potassium phosphate, 100 mM NaCl at pH 7), indicating a favorable electrostatic contribution of the guanidinium–carboxylate interaction involving the Arg20 and Glu18 side chains. The mutation designed to increase the solvation energy or hydrophobic interaction (L30Y) exhibited a more pronounced stabilizing effect in high salt, consistent with the effect of salt concentrations on increasing the hydrophobic effect (Jaenicke 2000). This effect could be the result of an extremely unfavorable denatured state wherein the hydrophobic side chains are exposed to aqueous solution where the high salt concentration strongly disfavors solvation. Not much variation in the stabilization derived from the mutation D34T was observed as a function of the salt concentration, as one would expect. The proteins with multiple mutations exhibit the expected balance between increased hydrophobic and decreased electrostatic contributions under high salt conditions.
Fluorescence-monitored GdnHCl denaturation
On the basis of the existence of a near-UV CD spectrum, our laboratory previously reported that the wild-type hYap WW domain is unfolded by chemical denaturants such as urea and GdnHCl to a non-random-coiled state, as opposed to the random-coiled denatured state afforded by thermal denaturation (no near-UV CD signal) (Koepf et al. 1999b). This behavior is exhibited by all the WW domain variants studied within, as is demonstrated in Figure 7A ▶ for the A20R/L30Y/D34T triple mutant. Most of the thermostable variants discussed in this paper did not entirely lose their tertiary structure in 8 M urea; therefore, the stronger denaturant GdnHCl was employed. Tryptophan fluorescence was monitored to probe the environmental changes around the tryptophans and hence protein structural changes. When excited at 295 nm, the native state WW domain showed maximum emission wavelength (λmax) around 343 nm. When we added 6 M GdnHCl, the λmax red-shifted to 358 nm with a decrease in intensity, indicating increased exposure of the Trp residues to the surrounding solvent (Fig. 7B ▶). Fluorescence-based denaturation curves (GdnHCl) recorded at 4°C are shown in Figure 7C ▶. The designed WW domain variants showed different fluorescence intensity (345 nm) at both 0 and 6 M GdnHCl relative to that of the wild type (Fig. 7C ▶), an indication of the different environment around the Trp residues in the proteins, in both the folded and the chemically denatured states. Given that the denatured states afforded by chaotrope and thermal denaturation may be different (Fig. 7A ▶), the results shown in Table 2 (fitted to a two-state model using equation 1) should be considered only as an estimate for assessing stability. The chaotrope-derived stability may or may not be directly comparable to the thermal stability, depending on the extent of the differences in the denatured states, which are the subject of ongoing studies (Table 1a). Table 2 indicates that the designed WW variants also exhibit increased stability, with the triple mutant (A20R/L30Y/D34T) being the most stable.
Fig. 7.



GdnHCl denaturation of hYap wild-type and variant WW domains. (A) Near-UV CD scans of A20R/L30Y/D34T in pH 7 buffer (4°C) (·), 6 M GdnHCl (○), and at 100°C (×). (B) Fluorescence emission scans of A20R/L30Y/D34T in buffer (solid line) and 6 M GdnHCl (dashed line). (C) GdnHCl denaturation curves determined by Trp fluorescence emission at 345 nm (4°C) with samples excited at 295 nm. The identities of the curves are as follows: (·) wild-type; (|ap) L30Y; (×) D34T; (○) A20R; (♦) A20R/D34T; (▴) A20R/L30Y; and (▪) A20R/L30Y/D34T. Solid lines are fitted to equation 1.
Table 2.
Stability of hYap wild-type and WW domain variants to GdnHCl denaturation
| WW sequence | ΔGu (H2O) (4°C) (kcal/mol) | ΔΔGu (H2O) (4°C) (kcal/mol) | m (4°C) (kcal/mol/M) |
| Wild type | 0.71 ± 0.10a | — | 1.12 ± 0.03 |
| L30Y | 1.29 ± 0.30a | 0.58 | 1.12 ± 0.11 |
| D34T | 1.15 ± 0.08a | 0.44 | 1.09 ± 0.02 |
| A20R | 1.58 ± 0.04 | 0.87 | 1.04 ± 0.02 |
| A20R/D34T | 1.88 ± 0.04 | 1.17 | 0.96 ± 0.01 |
| A20R/L30Y | 2.47 ± 0.04 | 1.76 | 0.97 ± 0.01 |
| A2R/L30Y/D34T | 3.17 ± 0.07 | 2.46 | 0.92 ± 0.02 |
a Higher error is expected for data fits with little or no pretransition baseline (Fig. 7C ▶).
Ligand binding
The wild-type hYap WW domain binds to proline-rich ligands with the consensus sequence PPXY; therefore, the following peptide EYPPYPPPPYPSG was employed as the ligand in this study (Macias et al. 1996; Verdecia et al. 2000). Because ligand binding to the WW domain involves burying Trp39 in the second hydrophobic cluster, the WW–ligand complex exhibits a slight blue shift in its Trp fluorescence emission maximum as well as a significant increase in the fluorescence intensity. These features can be conveniently used to monitor ligand binding (Koepf et al. 1999a). Although the D34T residue is on the opposite face of the sheet from the ligand-binding site, both the A20R and L30Y mutations are on the ligand-binding face. NMR studies reveal important contacts between the side chain of Leu30 and the conserved Tyr residue in the ligand (Macias et al. 1996). Therefore it is expected that a mutation at this position will affect binding (Espanel and Sudol 1999). Indeed, the L30Y mutation abolishes ligand binding (Fig. 8 ▶), in the physiological concentration range. All the other engineered thermostable hYap WW variants exhibit similar to slightly higher dissociation constants—3.1 ± 0.1 μM (D34T), 4.9 ± 0.2 μM (A20R), and 5.8 ± 0.2 μM (A20R/D34T)—in comparison to that of the wild type (2.5 ± 0.1 μM). The increased stability of the variants does not result in higher ligand-binding affinity. As a matter of fact, we discovered an hYap sequence that is largely unfolded and exhibits a dissociation constant similar to the folded wild-type WW domain (Koepf et al. 1999a). In addition, phage display generation of hYap variants that can bind the ligand identified WW domains that have non-cooperative thermal transitions, indicating the lack of conformational homogeneity (Dalby et al. 2000).
Fig. 8.

Peptide ligand (EYPPYPPPPYPSG) binding to the hYap wild-type and variant WW domains was monitored by fluorescence emission intensity at 345 nm. The ligand (1 mM) was titrated into wild-type (·), L30Y (|ap), D34T (×), A20R (○), and A20R/D34T (♦) WW domains at a concentration of 9 μM. Solid lines are fits to equation 6.
Discussion
Most homology-based protein design efforts have focused on replacing poorly conserved residues at a given position with the most representative (or the consensus) type of residue. Stabilizing mutations discovered through trial and error are typically combined to generate "super-stable" proteins, whereas destabilizing point mutations are no longer considered. Herein, a slightly different approach is used in which the focus is on semi-conserved core residues (defined as residues that exhibit ≥50% consensus yet are not highly conserved) and their immediate structural neighbors. By combining both sequence and structural information, stabilizing single mutations and multiple mutations were identified with a high success rate.
Stabilizing forces provided by the mutations
The three mutations—D34T, A20R, and L30Y—were envisioned to stabilize the hYap WW domain by using different mechanisms. D34T possibly acts to disrupt the dipole attraction between the side chains of Asn31 and Asp34, allowing Asn31 to form what we expect to be two or three more hydrogen bonds with the protein backbone, by analogy with the Pin WW domain. Because Asn31 resides at the end of the central β-strand, it is likely to be able to reach out to the protein backbone of both the first strand and the loop between the second and the third strands, analogous to Asn26 in Pin (Fig. 2B ▶).
Another possible stabilizing effect provided by the D34T mutation relates to the role this residue plays in stabilizing a π-turn. The conformation of residues His32 to Gln35 approximate a common π-turn motif consisting of an αr-α r-αr-αL loop ((Sibanda and Thornton 1985; Thornton et al. 1988; Efimov 1993; Gunasekaran et al. 1997; DeGrado et al. 1999). The first three residues of the loop form almost a full turn of an α-helix. As a result, the residue that proceeds this turn has a preference for side chains that can form a classical N-cap motif, such as Asn31 in hYap WW domain. However, Asp is the most preferred residue based on studies of the B1 IgG-binding domain (Zhou et al. 1996). This study also indicated a high frequency of occurrence of a Ser or Thr residue at position 3 in the π-turn (Zhou et al. 1996), corresponding to residue Asp34 in the wild-type hYap. The hydroxyl group from Ser or Thr generally accepts a hydrogen bond from an amide two residues in the C-terminal direction in sequence (thereby stabilizing the intervening residue in an αL conformation) while simultaneously forming a hydrogen bond to the N-capping residue. Therefore, it is also possible that the D34T mutation in hYap stabilizes the native structure of the protein simply because Thr is intrinsically favored over Asp at this position.
The D34T mutation in the hYap WW domain increased the Tm by 5°C and ΔGu by 0.38 kcal mol−1 at 65°C (thermal unfolding) or 0.44 kcal mol−1 at 4°C (GdnHCl denaturation). Such an increase in stability could be the result of the putative importance of hydrogen bonding between a side chain and the protein backbone (Strop and Mayo 2000), or π-turn forming propensity at this position as suggested by a genetic and structural analysis (Zhou et al. 1996).
The surface mutation, A20R, stabilizes the protein both by increasing favorable electrostatic interactions with a nearby carboxylate group and by burying hydrophobic surface against the neighboring Leu30 side chain (Fig. 4 ▶). Generally, placing charged groups on the surface of folded proteins to increase stability has been attributed to either improved electrostatic interactions in the folded state or the influence of charge on the ensemble of denatured states (Pace et al. 2000). In this case, we believe that the A20R mutation provides stabilizing forces beyond favorable electrostatic interactions in the folded state. There are at least two pieces of evidence to support this statement. First, the stabilizing effect of A20R was mostly retained in 2 M NaCl, indicating the existence of more substantial stabilizing forces than simply electrostatic interactions (Table 1). Furthermore, a calculated increase in the solvent-buried hydrophobic surface area was observed for Leu30 and other hydrophobic residues in the second cluster, owing to the A20R mutation, which further contributes to solvation energy. Together, favorable electrostatic and hydrophobic interactions seem to explain the increase of 0.89 kcal mol−1 (65°C) or 0.87 kcal mol−1 (GdnHCl, 4°C) in thermodynamic stability and the 11.4°C increase in Tm relative to the wild type.
In the wild-type hYap WW domain structure, the side chain of Leu30 is partially exposed in the central strand, surrounded by the side chains of Glu18, Ala20, Tyr28, His32, and Thr37 in hydrophobic cluster 2. The L30Y mutation contributed only moderately to stability when the wild-type hYap WW domain was used as the host because the Leu → Tyr mutation only slightly increased the buried hydrophobic surface area of the protein, as confirmed by the small (2.9°C) increase in Tm for the L30Y mutant. However, when A20R hYap variant was used as the host, the Tyr side chain at position 30 becomes more extensively buried owing to the complimentary hydrophobic surface created by the extended Arg20 side chain. It is also possible that a highly favorable His-aromatic interaction between Tyr30 and His32 could contribute to stabilization in the folded state. The L30Y mutation introduced in the context of A20R WW sequence results in 40 Å2 increase in ΔASA at position 30 alone, which should contribute favorably to both the ΔGu and the Tm. Overall, the A20R/L30Y mutations raised the Tm of hYap WW domain by >22°C, the ΔGu by 1.86 kcal mol−1 (65°C), and 1.76 kcal mol−1 (4°C, GdnHCl) relative to the wild type. This increase in thermodynamic stability is beyond the additive effects of both mutations as a result of molecular recognition between the two newly introduced side chains. It is worth pointing out that an A20Y/L30Y variant showed a 12°C increase in Tm (Ibragimova and Wade 1999), as opposed to the 22°C increase for the A20R/L30Y variant discussed here, underscoring the importance of favorable electrostatic interactions brought about by the Ala to Arg mutation.
Because the D34T and A20R/L30Y stabilizing mutations are spatially segregated in the three-dimensional structure of the WW domain, making all three mutations in the same sequence should further stabilize the hYap WW domain in an additive fashion. Indeed, the A20R/L30Y/D34T variant has a Tm of 79.6°C (28°C higher than that of the wild-type protein), and a ΔΔGu of 2.54 kcal mol−1(65°C) or 2.46 kcal mol−1(4°C, GdnHCl) relative to the wild type. The fact that the relatively small changes in free energy give rise to such significant changes in Tm for this protein is not surprising, considering its small size (57 residues) and correspondingly low ΔH and ΔCP (Alexander et al. 1992).
Notably, the Tm (79.6°C) of the triple mutant hYap WW domain (A20R/L30Y/D34T) has far surpassed the stability of the FBP28 (Tm = 62°C) and Pin (Tm = 58°C) WW domains, on which most of the structural homology was based. This could be partly the result of the fact that the hYap WW domain under study is ∼20–25 residues longer than the other two WW domain proteins. Several shorter versions of the hYap wild-type proteins were shown to be less stable (E. Koepf and J.W. Kelly, unpubl.). The two termini of the hYap protein meet on top of and cover the first hydrophobic cluster. This structural arrangement helps exclude solvent molecules. Although WW domains with fewer residues have their termini in close proximity, they do not provide as much shielding to the first hydrophobic cluster and therefore are likely to have lower stability in comparison to the 57-residue A20R/L30Y/D34T variant of the hYap WW domain.
The NMR spectra (Fig. 5 ▶) reveal that all the stabilizing variants when combined exhibit more dispersed chemical shifts relative to wild-type hYap or single-site variants thereof, indicating subtle changes in protein structure and/or dynamics. For example, the Trp17 indole N-H in the A20R/L30Y/D34T WW variant has a proton chemical shift of 10.68 ppm, a 0.2-ppm downfield shift in comparison to that of the wild-type protein. Because Trp17 was shown previously to be essential for hYap WW domain folding (Koepf et al. 1999a), one should not be surprised to see a correlation between a better-structured and more extensive hydrophobic core around Trp17 and increased stability.
Conclusion
This study shows that the combination of sequence homology and structural alignment within a given protein family can be used to increase stability by judicious choice of mutations. Here, we used the simplest three-stranded β-sheet, the WW domain, to demonstrate that it is possible to make a triple mutant of the hYap domain (A20R/L30Y/D34T), which acts on two isolated regions of the protein to increase thermal stability by 28°C. The A20R/L30Y mutations act synergistically to increase stability by an electrostatic interaction and through complimentary hydrophobic interactions, whereas the D34T mutation may either allow the N31 side chain to make two to three additional hydrogen bonds to the backbone and/or, alternatively, stabilize a π-turn. The advantage of using structural information within a protein family is that it enables consideration of important side-chain interactions in the context of the protein structure (Thornton et al. 1993; Babbitt and Gerlt 2001), therefore allowing a much higher success rate in protein design than simply considering sequence homology. Overall, this approach presents a simple and effective way to engineer stable WW domains and is complementary to phage display or other combinatorial methods used to randomize portions of the sequence in search of stable or functional variants (Dalby et al. 2000; Toepert et al. 2001).
Materials and methods
All chemicals used were of reagent grade. The exact concentration of GdnHCl was determined by reflective index (Krivacic and Urry 1971).
Protein preparation
The sequence of the wild-type 57-residue hYap WW domain construct used for this study is GSMSFEIPDDVPLPAGWEMAKTSS GQRYFLNHIDQTTTWQDPRKAMLSQMNVTAPTS. All mutant DNAs were prepared with the QuickChange site-directed mutagenesis procedure from Stratagene (La Jolla, CA), using wild-type DNA as the template; the mutations were verified by DNA sequencing. The wild-type hYap WW domain and the mutants were expressed recombinantly in Escherichia coli and purified as described previously (Koepf et al. 1999b). Protein sequence integrity was confirmed by electro-spray ionization mass spectrometry, and purity was evaluated by analytical HPLC. The concentrations of all protein solutions were determined by absorbance at 280 nm, using an ɛ = 11,000 M−1 cm−1 for wild-type, A20R, and D34T, and an ɛ = 12,000 M−1 cm−1 for WW domains with the L30Y mutation. A standard buffer containing 20 mM potassium phosphate (pH 7), 100 mM NaCl, 0.02% NaN3 was used for all measurements, except those in high salt, for which 2 M NaCl was used.
Fluorescence-monitored denaturation
Fluorescence measurements were performed on an Aviv Model ATF105 Automated Titrating Differential/Ratio Spectrofluorometer (Lakewood, NJ), using an automated titrator. All samples were excited at 295 nm with a bandwidth of 2 nm, whereas fluorescence emission was measured at 345 nm with a bandwidth of 6 nm. Sample solutions were stirred vigorously for 4 min after each addition of titrant by the instrument to ensure that the samples reached equilibrium. GdnHCl denaturation studies were conducted at 4°C . The WW domain samples (2 μM) in the standard buffer were titrated with 2 μM protein in 8 M GdnHCl, maintaining a constant volume of 2 mL. A two-state model was invoked with each WW domain studied to obtain free energy of unfolding (ΔGu) (see Results) by using equation 1 (Pace and Scholtz 1997):
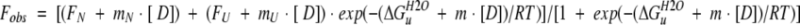 |
1 |
where Fobs is the observed fluorescence emission intensity at 345 nm, ΔGuH2O is the free energy of unfolding in water (by extrapolation), and m measures the dependence of ΔGu on denaturant concentration. The slope of the pretransition/posttransition baselines are given by mN and mU, respectively, whereas the intercepts of the baselines are given by FN and FU, respectively.
Circular dichroism spectroscopy
All circular dichroism (CD) experiments were conducted on an AVIV Model 202SF Stopped Flow Circular Dichroism Spectrometer equipped with a Peltier temperature-controlled cell holder. WW domains (50 μM) in a 0.1-cm pathlength Suprasil quartz cuvette (Hellma, Forest Hills, NY) were used for far-UV thermal denaturation studies. Samples were heated/cooled with 2°C steps (10°C/min) with a 2-min equilibration time at each temperature before data acquisition. Ellipticity change as a function of temperature was fitted according to equations 2 and 3:
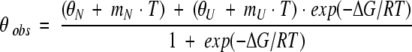 |
2 |
in which
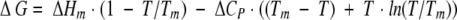 |
3 |
where θobs is the observed ellipticity at temperatures T (Kelvin), mN and mU are the slopes of the pretransition and posttransition regions, respectively, of which the intercepts are represented by θN and θU, respectively. ΔCP is the heat capacity, and ΔHm is the enthalpy at the unfolding transition with a midpoint of Tm. The fraction of unfolded protein (α) at each temperature is calculated using equation 4:
 |
4 |
and the ΔGu at 65°C (338°K) was calculated based on the fraction unfolded (α) by using equation 5:
 |
5 |
1H NMR Spectroscopy
All 1H NMR spectra were recorded on an AMX 600 MHz spectrometer. 3-(Trimethylsilyl) propionate-2,2,3,3,-d4 was used as internal chemical shift standard. Typically, spectra were acquired at 25°C at a concentration of 150 μM (pH 7). Water suppression was achieved using the Watergate pulse sequence.
Energy simulations
Molecular modeling was performed on an SGI Octane workstation, using the software package Insight II (Molecular Simulations, Inc., San Diego, CA). Energy calculations were performed using the Discover module within Insight II. Solvent-accessible surface area was calculated using the Connolly algorithm with a 1.4 -Å radius water probe (Connolly 1983).
Ligand binding
The proline-rich ligand EYPPYPPPPYPSG was synthesized using standard solid-phase peptide synthesis and purified via HPLC (Koepf et al. 1999a). Binding of the ligand to the hYap WW domain and variants thereof was measured using an automated titrator on an Aviv Model ATF105 Spectrofluorometer. Peptide ligand (1 mM) dissolved in buffer was titrated into a cuvette containing 2 mL of a 9 μM WW domain solution. Samples were vigorously stirred for 3 min after each addition of the ligand to enable equilibrium to be reached. Fluorescence emission was monitored at 345 nm by exciting the samples at 295 nm. After background subtraction, the emission intensity at 345 nm (Fobs) was plotted as a function of total ligand concentration added in the sample (Ltotal) and fitted according to equation 6:
 |
6 |
where Ffree and Fbound are the fluorescence intensity without ligand and with ligand fully bound, respectively. P0 is the protein concentration in the solution, while Kd is the dissociation constant associated with interaction of the WW domain and ligand.
Acknowledgments
The authors are grateful to the NIH (GM51105) for primary financial support and the Lita Annenberg Hazen family as well as the Skaggs Institute of Chemical Biology for additional support. We are grateful to Dr. W.F. DeGrado for critical reading of the manuscript. We thank Dr. E. Koepf for his help during the initial stages of this project, Dr. M. Jäger for helpful discussions, and S. Deechongkit for his help with mass spectrometry and ultracentrifugation studies.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10. 1101/
References
- Alexander, P., Fahnestock, S., Lee, T., Orban, J., and Bryan, P. 1992. Thermodynamic analysis of the folding of the streptococcal protein G IgG-binding domains B1 and B2: Why small proteins tend to have high denaturation temperatures. Biochemistry 31 3597–3603. [DOI] [PubMed] [Google Scholar]
- Babbitt, P.C. and Gerlt, J.A. 2001. New functions from old scaffolds: How nature reengineers enzymes for new functions. Adv. Protein Chem. 55 1–28. [DOI] [PubMed] [Google Scholar]
- Baker, D. 2000. A surprising simplicity to protein folding. Nature 405 39–42. [DOI] [PubMed] [Google Scholar]
- Bashford, D., Chothia, C., and Lesk, A.M. 1987. Determinants of a protein fold. Unique features of the globin amino acid sequences. J. Mol. Biol. 196 199–216. [DOI] [PubMed] [Google Scholar]
- Bedford, M.T., Chan, D.C., and Leder, P. 1997. FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J. 16 2376–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia, C., Gelfand, I., and Kister, A. 1998. Structural determinants in the sequences of immunoglobulin variable domain. J. Mol. Biol. 278 457–479. [DOI] [PubMed] [Google Scholar]
- Connolly, M.L. 1983. Solvent-accessible surfaces of proteins and nucleic acids. Science 221 709–713. [DOI] [PubMed] [Google Scholar]
- Crane, J.C., Koepf, E.K., Kelly, J.W., and Gruebele, M. 2000. Mapping the transition state of the WW domain β-sheet. J. Mol. Biol. 298 283–292. [DOI] [PubMed] [Google Scholar]
- Dahiyat, B.I., Sarisky, C.A., and Mayo, S.L. 1997. De novo protein design: Towards fully automated sequence selection. J. Mol. Biol. 273 789–796. [DOI] [PubMed] [Google Scholar]
- Dalby, P.A., Hoess, R.H., and DeGrado, W.F. 2000. Evolution of binding affinity in a WW domain probed by phage display. Protein Sci. 9 2366–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGrado, W.F., Wasserman, Z.R., and Lear, J.D. 1989. Protein design, a minimalist approach. Science 243 622–628. [DOI] [PubMed] [Google Scholar]
- DeGrado, W.F., Summa, C.M., Pavone, V., Nastri, F., and Lombardi, A.1999. De novo design and structural characterization of proteins and metalloproteins. Ann. Rev. Biochem. 68 779–819. [DOI] [PubMed] [Google Scholar]
- Desjarlais, J.R. and Handel, T.M. 1995. De novo design of the hydrophobic cores of proteins. Protein Sci. 4 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill, K.A. 1997. Additivity principles in biochemistry. J. Biol. Chem. 272 701–704. [DOI] [PubMed] [Google Scholar]
- Efimov, A.V. 1993. Patterns of loop regions in proteins. Curr. Opin. Struct. Biol. 3 379–384. [Google Scholar]
- Espanel, X. and Sudol, M. 1999. A single point mutation in a group I WW domain shifts its specificity to that of group II WW domains. J. Biol. Chem. 274 17284–17289. [DOI] [PubMed] [Google Scholar]
- Gunasekaran, K., Ramakrishnan, C., and Balaram, P. 1997. β-hairpins in proteins revisited: Lessons for de novo design. Protein Eng. 10 1131–1141. [DOI] [PubMed] [Google Scholar]
- Hecht, M.H., Richardson, J.S., Richardson, D.C., and Ogden, R.C. 1990. De novo design, expression, and characterization of felix: A four-helix bundle protein of native-like sequence. Science 249 884–891. [DOI] [PubMed] [Google Scholar]
- Huang, X., Poy, F., Zhang, R., Joachimiak, A., Sudol, M., and Eck, M.J. 2000. Structure of a WW domain containing fragment of dystrophin in complex with β-dystroglycan. Nat. Struct. Biol. 7 634–638. [DOI] [PubMed] [Google Scholar]
- Ibragimova, G.T. and Wade, R.C. 1999. Stability of the β-sheet of the WW domain: a molecular dynamics simulation study. Biophys. J. 77 2191–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenicke, R. 2000. Stability and stabilization of globular proteins in solution. J. Biotechnol. 79 193–203. [DOI] [PubMed] [Google Scholar]
- Jiang, X., Farid, H., Pistor, E., and Farid, R.S. 2000. A new approach to the design of uniquely folded thermally stable proteins. Protein Sci. 9 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, B.K., Williamson, M.P., and Sudol, M. 2000. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 14 231–241. [PubMed] [Google Scholar]
- Koehl, P. and Levitt, M. 1999. De novo protein design. I. In search of stability and specificity. J. Mol. Biol. 293 1161–1181. [DOI] [PubMed] [Google Scholar]
- Koepf, E.K., Petrassi, H.M., Ratnaswamy, G., Huff, M.E., Sudol, M., and Kelly, J.W. 1999a. Characterization of the structure and function of W to F WW domain variants: Identification of a natively unfolded protein that folds upon ligand binding. Biochemistry 38 14338–14351. [DOI] [PubMed] [Google Scholar]
- Koepf, E.K., Petrassi, H.M., Sudol, M., and Kelly, J.W. 1999b. WW: An isolated three-stranded antiparallel β-sheet domain that unfolds and refolds reversibly; evidence for a structured hydrophobic cluster in urea and GdnHCl and a disordered thermal unfolded state. Protein Sci. 8 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P. 1991. Molscript: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Krivacic, J.R. and Urry, D.W. 1971. Ultraviolet refractive indices of aqueous solutions of urea and guanidine hydrochloride. Anal. Chem. 43 1508–1510. [DOI] [PubMed] [Google Scholar]
- Larson, S.M. and Davidson, A.R. 2000. The identification of conserved interactions within the SH3 domain by alignment of sequences and structures. Protein Sci. 9 2170–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesk, A.M. and Fordham, W.D. 1996. Conservation and variability in the structures of serine proteinases of the chymotrypsin family. J. Mol. Biol. 258 501–537. [DOI] [PubMed] [Google Scholar]
- Lumb, K.J. and Kim, P.S. 1995. A buried polar interaction imparts structural uniqueness in a designed heterodimeric coiled coil. Biochemistry 34 8642–8648. [DOI] [PubMed] [Google Scholar]
- Macias, M.J., Hyvonen, M., Baraldi, E., Schulta, J., Sudol, M., Saraste, M., and Oschkinat, H. 1996. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature 382 646–649. [DOI] [PubMed] [Google Scholar]
- Macias, M.J., Gervais, V., Civera, C., and Oschkinat, H. 2000. Structural analysis of WW domains and design of a WW prototype. Nat. Struct. Biol. 7 375–379. [DOI] [PubMed] [Google Scholar]
- Maxwell, K.L. and Davidson, A.R. 1998. Mutagenesis of a buried polar interaction in an SH3 domain: Sequence conservation provides the best prediction of stability effects. Biochemistry 37: 16172–16182. [DOI] [PubMed] [Google Scholar]
- Michnick, S.W. and Shakhnovich, E. 1998. A strategy for detecting the conservation of folding-nucleus residues in protein superfamilies. Fold Des. 3 239–251. [DOI] [PubMed] [Google Scholar]
- Murphy, K.P. and Freire, E. 1992. Thermodynamics of structural stability and cooperative folding behavior in proteins. Adv. Protein Chem. 43 313–361. [DOI] [PubMed] [Google Scholar]
- Nikolova, P.V., Henckel, J., Lane, D.P., and Fersht, A.R. 1998. Semirational design of active tumor suppressor p53 DNA binding domain with enhanced stability. Proc. Natl. Acad. Sci. 95 14675–14680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N. 2001. Polar group burial contributes more to protein stability than nonpolar group burial. Biochemistry 40 310–313. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. and Scholtz, J.M. 1997. Measuring the conformational stability of a protein. In Protein structure, 2nd ed. (ed. T.D. Creighton), pp. 299–321. IRL Press, Oxford, UK.
- Pace, C.N., Alston, R.W., and Shaw, K.L. 2000. Charge–charge interactions influence the denatured state ensemble and contribute to protein stability. Protein Sci. 9 1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl, D., Mueller, U., Heinemann, U., and Schmid, F.X. 2000. Two exposed amino acid residues confer thermostability on a cold shock protein. Nat. Struct. Biol. 7 380–383. [DOI] [PubMed] [Google Scholar]
- Ranganathan, R., Lu, K.P., Hunter, T., and Noel, J.P. 1997. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 89 875–886. [DOI] [PubMed] [Google Scholar]
- Rufino, S.D. and Blundell, T.L. 1994. Structure-based identification and clustering of protein families and superfamilies. J. Comput. Aided Mol. Des. 8 5–27. [DOI] [PubMed] [Google Scholar]
- Schultz, J., Milpetz, F., Bork, P., and Ponting, C.P. 1998. SMART, a simple modular architecture research tool: Identification of signalling domains. Proc. Natl. Acad. Sci. 95 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, J., Copley, R.R., Doerks, T., Ponting, C.P., and Bork, P. 2000. SMART: A Web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 28 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhnovich, E.I. and Gutin, A.M. 1993. A new approach to the design of stable proteins. Protein Eng. 6 793–800. [DOI] [PubMed] [Google Scholar]
- Sibanda, B.L. and Thornton, J.M. 1985. β-Hairpin families in globular proteins. Nature 316 170–176. [DOI] [PubMed] [Google Scholar]
- Street, A.G. and Mayo, S.L. 1999. Computational protein design. Structure 7 R105–R109. [DOI] [PubMed] [Google Scholar]
- Strop, P. and Mayo, S.L. 2000. Contribution of surface salt bridges to protein stability. Biochemistry 39 1251–1255. [DOI] [PubMed] [Google Scholar]
- Sudol, M. 1996. Structure and function of the WW domain. Prog. Biophys. Mol. Biol. 65 113–132. [DOI] [PubMed] [Google Scholar]
- Sudol, M. and Hunter, T. 2000. New wrinkles for an old domain. Cell 103 1001–1004. [DOI] [PubMed] [Google Scholar]
- Thornton, J.M., Sibanda, B.L., Edwards, M.S., and Barlow, D.J. 1988. Analysis, design and modification of loop regions in proteins. BioEssays 8 63–69. [DOI] [PubMed] [Google Scholar]
- Thornton, J.M., MacArthur, M.W., McDonald, I.K., Jones, D.T., Mitchell, J.B.O., Nandi, L., Price, S.L., and Zvelebil, M.J.J.M. 1993. Protein structures and complexes: What they reveal about the interactions that stabilize them. Philos. Trans. R. Soc. London, Ser. A 345 113–129. [Google Scholar]
- Toepert, F., Pires, J.R., Landgraf, C., Oschkinat, H., and Schneider-Mergener, J. 2001. Synthesis of an array comprising 837 variants of the hYap WW protein domain. Angew. Chem. Int. Ed. Eng. 40 897–900. [DOI] [PubMed] [Google Scholar]
- Verdecia, M.A., Bowman, M.E., Lu, K.P., Hunter, T., and Noel, J.P. 2000. Structural basis for phosphoserine–proline recognition by group IV WW domains. Nat. Struct. Biol. 7 639–643. [DOI] [PubMed] [Google Scholar]
- Wang, Q., Buckle, A.M., Foster, N.W., Johnson, C.M., and Fersht, A.R. 1999. Design of highly stable functional GroEL minichaperones. Protein Sci. 8 2186–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells, J.A. 1990. Additivity of mutational effects in proteins. Biochemistry 29 8509–8517. [DOI] [PubMed] [Google Scholar]
- Zhou, H.X., Hoess, R.H., and DeGrado, W.F. 1996. In vitro evolution of thermodynamically stable turns. Nat. Struct. Biol. 3 446–451. [DOI] [PubMed] [Google Scholar]
- Zhu, H., Celinski, S.A., Scholtz, J.M., and Hu, J.C. 2000. The contribution of buried polar groups to the conformational stability of the GCN4 coiled coil. J. Mol. Biol. 300 1377–1387. [DOI] [PubMed] [Google Scholar]