

Abstract
Kinetic properties of the dimeric enzyme dUTPase from Leishmania major were studied using a continuous spectrophotometric method. dUTP was the natural substrate and dUMP and PPi the products of the hydrolysis. The trypanosomatid enzyme exhibited a low Km value for dUTP (2.11 μM), a kcat of 49 s−1, strict Michaelis–Menten kinetics and is a potent catalyst of dUDP hydrolysis, whereas in other dUTPases described, this compound acts as a competitive inhibitor. Discrimination is achieved for the base and sugar moiety showing specificity constants for different dNTPs similar to those of bacterial, viral, and human enzymes. In the alkaline range, the Km for dUTP increases with the dissociation of ionizable groups showing pKa values of 8.8, identified as the uracil moiety of dUTP and 10, whereas in the acidic range, Km is regulated by an enzyme residue exhibiting a pKa of 7.1. Activity is strongly inhibited by the nucleoside triphosphate analog α-β-imido-dUTP, indicating that the enzyme can bind triphosphate analogs. The existence of specific inhibition and the apparent structural and kinetic differences (reflected in different binding strength of dNTPs) with other eukaryotic dUTPases suggest that the present enzyme might be exploited as a target for new drugs against leishmaniasis.
Keywords: dUTPase, dUDPase, uracil, Leishmania major, competitive inhibition, nucleotide metabolism
One of the main steps in the prevention of hazardous incorporation of uracil into DNA by DNA polymerase is the maintenance of a low ratio of dUTP/dTTP (Kornberg and Baker 1991). dUTP pyrophosphatase (dUTPase, EC 3.6.1.23) catalyzes the hydrolysis of dUTP to dUMP, a substrate for thymidylate synthase, and PPi (Bertani et al. 1961), removing dUTP from the dNTP pool and suppressing DNA fragmentation through excision repair which in turn leads to an increase in the rates of mutation and recombination (Tye et al. 1977).
dUTPase is widespread in nature and has been found in a variety of eukaryotic and prokaryotic organisms as well as in many viruses (Bjornberg et al. 1993; Bergman et al. 1994; Climie et al. 1994; McIntosh et al. 1994). The enzyme is essential for viability in Escherichia coli, (El-Hajj et al. 1988) and Saccharomyces cerevisiae (Gadsden et al. 1993) and is the target of certain cytotoxic drugs in human cancer cells (Zalud et al. 1994). These observations suggest that the activity is essential for DNA replication and cell division.
The three-dimensional structures of dUTPase from E. coli (Cedergen-Zeppezauer et al. 1992; Larsson et al. 1996a), feline inmunodeficiency virus (FIV) (Prasad et al. 1996), human (Mol et al. 1996), and equine infectious anemia virus (EIAV) (Dauter et al. 1999) have been determined and reveal a trimeric arrangement of identical subunits, with three active sites, formed by the contribution of residues from each subunit (Larsson et al. 1996; Mol et al. 1996).
Kinetic measurements of the hydrolysis of dUTP by dUTPase have been improved with time. Traditionally, the activity is measured by separation and quantification of radiolabeled reaction products by TLC (Tye et al. 1977) or by column chromatography (Hoffman 1987), however this method is slow, time-consuming, and discontinuous. The continuous method is based on the fact that hydrolysis of dUTP to dUMP and PPi is coupled to a pH and pMg-dependent release of protons, so the pH change can be used to follow the reaction. For this purpose, the use of the stopped-flow pH indicator technique has been extended to measure dUTPase reaction kinetic parameters by monitoring complete progress curves. This method has allowed for a detailed kinetic characterization of different dUTPases of distinct organisms like E. coli (Larsson et al. 1996b), Herpes simplex virus type I (HSV-I) (Bergman et al. 1995), and EIAV (Nord et al. 1997).
We have reported previously the presence in the parasitic protozoan Leishmania major of a cDNA encoding a functional dUTPase, isolated from a cDNA expression library by genetic complementation of dUTPase deficiency in E. coli. The gene is of single copy and has an open reading frame (ORF) encoding a protein of 269 amino acid residues, and a theoretical molecular mass of 30.3 kDa. So far, most of the dUTPases described have five amino acid consensus motifs in their sequences, however this parasitic dUTPase lacks these common motifs, and encodes a larger polypeptide (Camacho et al. 1997).
Recently, we have cloned the gene encoding the dUTPase into the expression vector pET-11c, and efficiently overexpressed the protein in E. coli. Recombinant dUTPase was purified using a combination of adsorption and anion-exchange chromatography. The enzyme proved to be a dimer in gel filtration and in cross-linking/SDS assays that represents the first time that a dimeric arrangement has been described for a dUTPase. Preliminary measurements using the discontinuous method showed that the enzyme hydrolyzes efficiently dUTP and dUDP into dUMP and PPi or Pi respectively, and is very sensitive to inhibition by dUMP, but not by PPi. The activity was highly dependent on the Mg2+ concentration (Camacho et al. 2000).
In this report we describe a detailed study of the kinetic properties of the enzyme, assessed by the stopped-flow spectrophotometric method. The results are discussed and compared to those reported for the trimeric dUTPases from E. coli (Larsson et al. 1996b), HSV-I (Bergman et al. 1995), the retroviruses of equine infectious anemia (Nord et al. 1997), and mouse mammary tumor virus (MMTV) (Bjornberg and Nyman 1996). Because of the marked differences between this enzyme and the rest of dUTPases described, a detailed study of the kinetic properties will provide valuable information that in the future may aid in the development of specific inhibitors.
Results
Kinetic characterization
Data of the multiple-turnover hydrolysis of dUTP by L. major dUTPase could be nicely fitted to the integrated Michaelis–Menten equation for enzymes with appreciable product inhibition, as demonstrated in Figure 1 ▶, obtaining these way values of Kmapp and Vmax, as described in Materials and Methods. Both of them remained constant with variations in the concentration of buffer/pH indicator. However, although the Vmax value was relatively insensitive to variations in substrate concentration (dUTP), there was an increase in Kmapp values with increasing dUTP concentrations, reflecting a process of product inhibition. After replotting the different values of Kmapp versus dUTP concentration, the real Km and the Kip value of product inhibition can be calculated (Fig. 2 ▶). Using dUTP concentrations between 10 and 300 μM, the real Km value obtained was 2.11 μM and Vmax was 35.98 ± 1.21 units mg−1. The hydrolysis product, dUMP, caused competitive inhibition with a Kip value of 13.05 μM. This low Kip value was confirmed by the addition of different concentrations of dUMP (5–20 μM) at t=0. Thus, different values of Kmapp were obtained and used for the analysis of competitive inhibition as described in Materials and Methods for α-β-imido-dUTP. The Kip value obtained using this method was 12.35 μM (Fig. 3 ▶). However, the other product of the reaction, PPi, did not inhibit the enzyme from L. (data not shown). Single turnover measurements gave a kcat value of 49 s−1. The specificity constant, kcat/Km for dUTP was thus 2.3 × 106 s−1M−1.
Fig. 1.

Complete hydrolysis of dUTP by dUTPase under multiple-turnover conditions. The inset shows the linear transformation of the data between the arrows, according to the integrated Michaelis–Menten equation adapted for enzymes with product inhibition as described in Materials and Methods, and the corresponding regression line. The reaction was recorded at 573 nm after reacting 30 nm dUTPase and 25 μM dUTP in the presence of 25 mM MgCl2, 2500 μM BICINE, and 50 μM cresol red at pH 8.
Fig. 2.

Replot of Kmapp vs. [S0]. Replot of the different slope values (Kmapp) from the linear plots of the inset of Figure 1 ▶, at different substrate concentrations from 10 to 300 μM, vs. [S]0. Following the integrated Michaelis–Menten equation for enzymes with product inhibition, the Kip value and the real Km could be obtained from this new linear replot as described in Materials and Methods.
Fig. 3.

Inhibition by dUMP. The Ki value for product (dUMP) inhibition was calculated with the Kmapp values obtained at different dUMP concentrations (5, 10, 15, and 20 μM using the formula shown in Materials and Methods for competitive inhibitors. The value obtained was 12.35 μM. The reactions were carried out using 50 μM dUTP and 30 nM dUTPase at pH 8 and 25°C.
dUDP appeared to be an efficient substrate for dUTPase with a Vmax of 99.38 ± 2.58 units mg−1. Analysis of the hydrolysis data of dUDP as described above for dUTP gave a real Km value of 62.70 μM, and a product inhibition constant (Kip) of 130.75 μM. The kcat for dUDP hydrolysis was calculated from the Vmax and the enzyme concentration, estimated from the extinction coefficient at 280 nm. Assuming that the enzyme was 100% active and that there are two active sites per dimer, the estimated kcat was 65 s−1 and the specificity constant, kcat/Km, was 1 × 106 s−1M−1.
PPi release
The release of pyrophosphate during the hydrolysis of dUTP was analyzed as described in Materials and Methods. A blank reaction, containing exactly the same volume as the sample to analyze and a control reaction, which was a standard sample of a known PPi concentration (16 μM), were analyzed. The control was used to calculate the apparent extinction coefficient of NADH. Samples with dUTP incubated 5 min in the presence of the enzyme were measured. The calculated PPi concentration was 24.71 ± 1.61 μM, which is equivalent to the initial dUTP concentration included in the reaction mixture suggesting that PPi and dUMP are the sole products of the reaction.
Effects of pH on Vmax and Km
The kinetic parameters for the hydrolysis of dUTP, Vmax and Kmapp, were determined in the pH interval 5.5–10, using different pairs of buffer/pH indicator. The Vmax value was relatively constant at pH 5.5–7 and 8.5–10, however there was a five-fold increase between pH 7–8.5 (Fig. 4a ▶). This increase in activity suggested the existence of a residue with pKa values in that range that may be important for catalysis. The corresponding values of Kmapp at different pH are shown in Figure 4b ▶ and imply that substrate binding is affected by at least three proton dissociations, according to the equation: 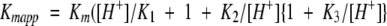 ). The best fit of the data to this equation was obtained for Km = 2.35 μM, revealing three pKa values for residues involved in substrate binding: pK1 = 7.1, pK2 = 8.8, and pK3 = 10. Titration of dUTP has been described to give a pKa of 8.9 which corresponds to the ionization of the uracil moiety (Dawson et al. 1986; Larsson et al. 1996a,b).
). The best fit of the data to this equation was obtained for Km = 2.35 μM, revealing three pKa values for residues involved in substrate binding: pK1 = 7.1, pK2 = 8.8, and pK3 = 10. Titration of dUTP has been described to give a pKa of 8.9 which corresponds to the ionization of the uracil moiety (Dawson et al. 1986; Larsson et al. 1996a,b).
Fig. 4.

Effect of pH on Km and Vmax. Data were obtained using different pH indicator/buffer pairs at 50/2500 μM at 25 °C. (a) Effect of pH on Vmax. (b) Effect of pH on Kmapp. Three pKa values (7.1, 8.8, and 10) were obtained by fitting the Km data to the equation: 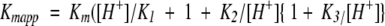 ), using a Km value of 2.35 μM. The concentration of MgCl2 was 25 mM.
), using a Km value of 2.35 μM. The concentration of MgCl2 was 25 mM.
Effects of temperature on Vmax and Km
Analysis of Vmax and Km values at different temperatures from 4°C to 60°C allowed us to conclude that the Km value is apparently insensitive to temperature variations. However, clear changes in Vmax occurred. From 4°C to 50°C there was an increase in activity, reaching a maximum at 50°C. At higher temperatures (≈60°C), dUTPase activity decreased very quickly, and in a short time was no longer detectable due to denaturation. In the interval from 4°C to 55°C, where denaturation is negligible, the increase of activity was exponential. Linear fitting of data to the Arrhenius equation (Cornish-Bowden et al. 1995) gave a value for the activation energy of 12,500 cal mol−1.
Metal ion requirement
In the absence of MgCl2, L. major dUTPase was totally inactive. The Vmax value increased with increasing concentrations of Mg2+ from 14 units mg−1 at 0.5 mM to 40 units mg−1 at 25 mM. This latter value remained constant at higher Mg2+ concentrations. No change in the apparent Km was observed when the Mg2+ concentration was raised from 0.5 to 250 mM.
No enzymatic activity was observed when Mg2+ was replaced by Ca2+ or Cu2+. However, the addition of Mn2+ or Co2+ instead of Mg2+ to the reaction mixture produces an increase in Km between 2- and 4-fold the value obtained with Mg2+, whereas Vmax remained unaltered.
The pH variation during the reaction was different depending on the metal ion used because the increase in the absorbance measured was different. This observation suggests that the nature of the ion used in the reaction has an important role in the number of protons released during the hydrolysis of dUTP.
Hydrolysis of other nucleotides
The hydrolysis of different nucleotides by L. major dUTPase was studied under the same conditions as used in the measurements of dUTP hydrolysis. The enzyme assay was carried out using as substrates dTTP, dCTP, dATP, and dGTP at a final concentration of 500 μM, and 0.3 μM of dUTPase. All nucleotides tested were inefficiently hydrolyzed compared to the substrates dUTP and dUDP. Table 1 shows the Kmapp and kcat values obtained with the different nucleotides. The rate of hydrolysis of UTP was extremely low; the Km value was higher than 2500 μM, and the specificity constant lower than 2 × 103 M−1s−1.
Table 1.
Catalytic parameters Km(μM), kcat(s−1) and specificity constant (M−1s−1) of Leishmania major dUTPase for different nucleoside triphosphates and UTP
| Substrate | Km | kcat | kcat/Km |
| dUTP | 2.11 | 49 | 2.3 × 107 |
| dATP | 1034 | 5.4 | 5.1 × 103 |
| dTTP | 1514 | 8.6 | 5.5 × 103 |
| dGTP | > 2000 | 6.4 | 6.4 × 103 |
| dCTP | > 2500 | 13 | 5.0 × 103 |
| UTP | > 2500 | <0.5 | < 2 × 103 |
Inhibition
We have characterized in detail strong competitive product inhibition of dUTPase by dUMP. Additionally, two substrate analogs have been tested as possible inhibitors of Leishmania dUTPase. First DMT-dU, an efficient inhibitor of E. coli dUTPase, was analyzed at concentrations from 10 to 1000 μM. Different Kmapp values were obtained, whereas the Vmax values were constant, indicating competitive inhibition with a Ki value higher than 1000 μM. Studies were also performed with α-β-imido-dUTP, using concentrations from 0.3–6 μM, which proved to be an effective inhibitor of the enzyme with a Ki value of 0.89 μM. (Fig. 5 ▶). Both Ki values were obtained taking into account product inhibition. No hydrolysis of these inhibitors could be detected after overnight incubations with 0.3 μM dUTPase.
Fig. 5.

Inhibition of Leishmania major dUTPase by α-β-imido-dUTP. Hydrolysis was performed using 30 nM enzyme at pH 8 and 25°C with (a) 25 μM dUTP and no inhibitor, and (b) 25 μM dUTP and 10 μM α-β-imido-dUTP. Inset linear dependence of apparent Km on inhibitor concentration.
Discussion
Protozoan dUTPases differ profoundly from general eukaryotic dUTPases in amino acid sequence and presumably structural features. Previous studies have been carried out with bacterial, viral, and human dUTPases (Bjornberg and Nyman 1996; Larsson et al. 1996a,b; Nord et al. 1997). These enzymes are homotrimers or monomers and are highly specific for dUTP. In the present study we perform an in-depth kinetic characterization with a dimeric trypanosomatid dUTPase and compare results with the corresponding data available at the moment for homotrimeric and monomeric dUTPases.
The kinetic constants for the hydrolysis of dUTP are similar to other dUTPases, with a low Km for dUTP, and a high specificity constant. Conversely, the estimated kcat for dUTP hydrolysis, obtained from single-turnover experiments, was higher than other dUTPases described so far. This kcat was two-fold higher than the kcat obtained using Vmax, which might be attributable to a "burst effect" caused by the high product (dUMP) inhibition, which slows down the reaction in multiple turnover experiments.
One of the main peculiarities of L. major dUTPase is the capacity of hydrolysis of dUDP. The Km was 30-fold higher, suggesting lower binding to the active site than in the case of the nucleoside triphosphate although the specificity constant was similar to the value obtained for dUTP. This characteristic is common only to the T2-T4 phage dCTPase-dUTPase, whereas dUDP is a strong competitive inhibitor for the rest of dUTPases with Ki values between 3 and 17 μM. In fact the E. coli, EIAV, and human dUTPases have been crystallized in complex with this nucleoside diphosphate (Larsson et al. 1996a,b; Prasad et al. 2000), which it is not hydrolyzed. The biological role of dUDPase activity may be to assure production of dUMP, the substrate of thymidylate synthase. It is also possible that intracellular levels of dUDP are high in these organisms and that this nucleotide is readily converted to dUTP, which is toxic to cells. Hydrolysis of dUDP would further ensure low dUTP levels and reduced incorporation of uracil into DNA.
Regarding the products of the reaction, the pyrophosphate activity measurements confirm that in the hydrolysis of dUTP, the products are dUMP and PPi. However, although dUMP acts as a strong competitive inhibitor, PPi does not inhibit the reaction as in other organisms (Larsson et al. 1996a,b), and the Kip value for dUMP is ∼10-fold lower in the case of the Leishmania enzyme. This observation might have interesting biological implications and further studies would be of interest to determine the possible influence of the dUMP pool and its utilization by thymidylate synthase on dUTPase activity.
The analysis of the influence of pH on Vmax suggests that the enzyme might have a residue important for activity with a pKa of ∼7–8. Conversely, the pH dependence of Km shows that formation of the enzyme-substrate complex requires deprotonation of a group exhibiting a pKa of about 7.1. Taking into account these observations, a general base catalysis mechanism for the hydrolysis of the substrate by dUTPase mediated by a histidine residue could be postulated. The dependence of Km also gives a pKa value of 10 suggesting that a tyrosine residue could be involved in substrate binding. There are several candidates that might correspond to the mentioned residues such as His-82 or Tyr-191 (Camacho et al. 1997). Thus, for instance Tyr-191 is present in a consensus sequence that appears in the uridine-binding site of enzymes such as pseudouridin synthetase (Kooning 1996). Site-directed mutagenesis is underway to confirm these observations.
The importance of the metal ion in the hydrolysis of dUTP has been demonstrated by the finding that metal-free dUTP is not hydrolyzed by E. coli dUTPase and that Mg2+ enhances the binding of dUTP by a factor of 100 (Larsson et al. 1996a,b). For instance, optimal activity values are reached at 50 mM of MgCl2 in the case of the enzymes of Bacillus subtilis and HSV (Price and Frato 1975). L. major dUTPase presents a maximum of activity at 25 mM of Mg2+ and almost the same Vmax values were obtained with Mn2+and Co2+ at the same concentration. With the latter ions, the Km values were 2- and 3-fold higher, respectively, suggesting that the metal ion might play an important role in the binding of dUTP to the active site, although as indicated for the human enzyme, this requirement could be purely structural (Mol et al. 1996). Accordingly, Ca2+ could not substitute Mg2+ in catalysis, probably attributable to the greater bulk of this ion, which can disturb the binding of the substrate to the active site, avoiding the hydrolysis. L. major dUTPase can discriminate easily between dUTP and the other common dNTPs, thus it is almost exclusively specific for dUTP, showing a specificity constant three orders of magnitude higher for this nucleotide than for the rest of substrates tested. The next best substrates were dATP and dTTP. Assuming that the association rate constant of enzyme and substrate is equal in bacterial, human, and viral dUTPases, and using the Km values for dUTP hydrolysis to normalize inhibitor binding by the different enzymes, the binding discrimination between dUTP and other dNTPs would be over 30 times greater in these dUTPases than in the case of the L. major dUTPase. The fact that this enzyme can discriminate not only the uracil moiety from other closely related bases, but also the deoxyribose moiety from ribose is essential to prevent a wasteful and possibly fatal hydrolysis of nucleotides needed for synthesis of DNA and RNA.
Our results show that Leishmania dUTPase has a slow turnover and an almost optimized specificity constant, characteristics which are common to all the dUTPases described to date. The similar kinetic properties suggest that the enzyme is equivalent in function and would refute the hypothesis that a slow turnover and good specificity constant are essential for an almost exclusive transfer of dUMP from dUTP.
The strong competitive inhibition properties of the triphosphate analog α-β imido-dUTP have been analyzed in different dUTPases like E. coli (Larsson et al. 1996b), HSV-1 (Nord et al. 1997), and EIAV (Nord 2000). As shown in Table 2, in the case of L. major dUTPase, the Ki value was considerably low, and in consequence the selectivity indexes relative to the rest of the eukaryotic enzymes were >10. These data suggest that a pronounced requirement for a triphosphate moiety to form a catalytic complex is necessary and provides support for the differences in the architecture of the active sites between dUTPases of trypanosomatids and other organisms. In addition, the lack of inhibition in the case of the Leishmania enzyme by the DMT-dU compared to the low Ki described for the E. coli enzyme (Persson et al. 1996), denotes possible structural differences between both dUTPases and the possibility of a specific inhibition of the parasitic enzyme.
Table 2.
Michaelis-Menten, specificity and inhibition constants of viral (EIAV, HSV-1 and MMTW), bacterial ( E. coli) and Leishmania major dUTPases towards different nucleoside triphosphates and uracil triphosphate analogues
| Substrate/inhibitor | L. major | E. colia | EIAVb | HSV-lc | MMTVd |
| dUTP: Km(kcat/Km) | 2.11 (2.3 × 107) | 0.2 (4 × 107) | 1.1 (2 × 107) | 0.3 (2 × 107) | 0.8 (2 × 106) |
| dTTP:Km(kcat/Km) | 1514 (5.5 × 103) | > 20000 (34) | 260 (< 2000) | 400 (1000) | N.D. (2000) |
| dCTP:Km(kcat/Km) | > 2500 (5.0 × 103) | 4000 (< 100) | 3000 (1000) | 1000 (2000) | N.D. |
| UTP:Km(kcat/Km) | > 2500 (< 2 × 103) | 2500 (< 100) | N.D. | 1000 (200) | N.D. |
| dUDP:Km(kcat/Km) | 62.70 (1 × 106) | Ki = 15 | Ki = 3.6 | Ki = 17 | N.D. |
| dUMP: Kip | 13.05 | 1500 | 130 | 170 | N.D. |
| α-β-imido-dUTP: Ki | 0.89 | 5 | 0.6 | 0.9 | N.D. |
The units for Km and Kiare μM and for kcat/Km M−1s−1. All values were obtained at pH 8.
a Larsson et al. 1996b.
b Nord et al. 1997.
c Bergman et al. 1995.
d Bjornberg and Nyman 1996.
In summary, dimeric L. major dUTPase does not exhibit any of the five consensus motifs present in the bacterial, viral, and human enzymes (Camacho et al. 2000) but displays comparable kinetic parameters for dUTP hydrolysis with marked dissimilarities in the binding of dUDP and dUMP, suggesting differences in the structure of the active sites. Diffractable material has been obtained in crystallization trials of T. cruzi dUTPase, another parasitic enzyme that displays a 55% identity with L. major (Bernier-Villamor et al. 1999). If structural information is made available, it may be exploited in the design of specific nucleotide analogs potentially useful for the medical treatment of leishmaniasis.
Materials and methods
Chemicals
2`-dUTP, 2`-dTTP, 2`-dCTP, and UTP were purchased from Pharmacia. Sodium salts of 2`-dCDP, 2`-dUDP, 2`-dUMP, 2`-dATP, and 2`-dGTP, (DMT-dU), the "pyrophosphate reagent," bovine albumin (BSA), CaCl2, MnCl2, MgCl2, CuCl2, CoCl2, and the pH indicators cresol red, alyzarin yellow, bromocresol purple, and bromothymol blue were from Sigma. The buffer N, N-bis (2-hydroxyethyl) glycine (BICINE) was obtained from USB. Recombinant enzyme was overexpressed and purified to homogeneity following the protocol described by Camacho et al. (2000) and stored in 50 mM BICINE at pH 8.0 and 25 mM MgCl2 at −80°C. The monodisperse and nonionic detergent octaethylene glycol mono-n-dodecylether (C12E8) was obtained from Nikko Chemicals. All the concentrations of the nucleotides and the enzyme were calculated spectrophotometrically (HP-8453, Hewlett Packard) at 280 nm, using the extinction coefficient (ɛ280 nm = 1.75 ml mg−1cm−1). The purity of the nucleotides was assessed by a MonoQ column attached to an FPLC (Pharmacia LKB), equilibrated with 0.01 M HCl in 0.01 M KCl and elution performed with a 20 ml gradient of 0.01 M to 0.2 M KCl in 0.01 HCl. Other chemicals used in these experiments were of the highest quality available.
Kinetic measurements
Nucleotide hydrolysis was monitored by mixing enzyme and substrate with a rapid kinetic accessory (Pharmacia) attached to a spectrophotometer (HP-8453, Hewlett Packard) and connected to a computer for data acquisition and storage. The measurements were performed at 25°C, and the solutions containing 25 mM MgCl2 and 1 mg ml−1 BSA or 0.01% C12E8 (w/v) were degassed previously. Protons, released through the hydrolysis of nucleotides, were neutralized by a pH indicator in a weak buffered medium with similar pKa and monitored spectrophotometrically at the absorbance peak of the basic form of the indicator. The ratio between the indicator and the buffer concentration was 50:2500 (μM), but it depended on the number of protons expected to be released in the reaction, the buffer capacity of the solution, and the extinction coefficient of the pH indicator. Absorbance changes were kept within 0.1 units. The indicator/buffer pairs were bromocresol purple/MES (pH 5.7–6.2, 605 nm), bromothymol blue/MOPS (pH 6.5–7.3, 622 nm), cresol red/BICINE (pH 7.5–8.5, 573 nm), and alyzarin yellow/CHES (pH 9.0–10.0, 445nm). Prior to each experiment, the apparent pKa values of each indicator/buffer pair were determined by titration, in the stopped-flow spectrophotometer, of solutions identical to those used in the kinetic experiments but without enzyme and substrate. The final enzyme concentration in multiple-turnover assays was 0.03 μM, 10–300 μM dUTP, 25 mM MgCl2 and the ionic strength 0.1 M, at pH 8. The majority of experiments were performed at 25°C. Indicator absorbance changes corresponding to complete hydrolysis of nucleotides were recorded in the computer, and the kinetic parameters Vmax and Kmapp (or slope) were calculated by fitting the data to the integrated Michaelis–Menten equation, adapted to enzymes which have an appreciable affinity for the product. The decrease in velocity with time is due to the decrease of saturation of the enzyme with substrate and the increase of the product inhibition: 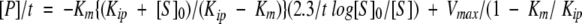 ), where
), where 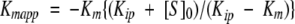 (Segel 1975). Data points in the region of equilibrium and the immediate start were omitted. To obtain the real Km for substrate hydrolysis and the Kip for product inhibition, the obtained slopes from the plots of the integrated velocity equation (or Kmapps obtained at different substrate concentrations) were replotted versus [S0]. Units for Vmax were μmol min−1.
(Segel 1975). Data points in the region of equilibrium and the immediate start were omitted. To obtain the real Km for substrate hydrolysis and the Kip for product inhibition, the obtained slopes from the plots of the integrated velocity equation (or Kmapps obtained at different substrate concentrations) were replotted versus [S0]. Units for Vmax were μmol min−1.
Single-turnover measurements were performed by reacting four-fold excess dUTPase (2–32 μM) with dUTP (0.5–8 μM), in the presence of cresol red (0.5 mM) and BICINE (25 mM), using the stopped-flow spectrophotometer at 25°C and a fixed concentration of 25 mM MgCl2. Instead of BSA we used the detergent C12E8, because of the interference in the absorbance at short times of BSA. The rate constant observed for the exponential absorbance traces are given by the equation kobs ≈ kcat/(1 + Km/Ce), as described by Larsson et al. (1996a,b). When the enzyme concentration (Ce) is at least four times the substrate concentration and Ce >> Km it is assumed that kobs ≅ kcat.
PPi release
The "pyrophosphate reagent" (Sigma, ref. P7275) for enzymatic determination of pyrophosphate, was utilized to quantify spectrophotometrically the amount of PPi produced in the hydrolysis of dUTP by dUTPase, as described by the manufacturer. The amount of pyrophosphate released was determined using a coupled enzyme system containing fructose-6-phosphatase kinase (pyrophosphate-dependent), aldolase, triosephosphatase isomerase and α-glycerophosphate dehydrogenase. Two moles of NADH are oxidized to NAD+ per mole of pyrophosphate consumed. The reaction is monitored spectrophotometrically at 340 nm. Measuring the increase of the absorbance of a blank containing only water, and the increase due to the PPi released after the incubation of 2 μM dUTPase with 25 μM dUTP at 30°C during 5 min, the amount of PPi in the sample can be calculated with the equation: PPi (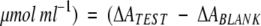 ) 4.82 (O'Brien 1976).
) 4.82 (O'Brien 1976).
Metal ion requirements
To study the metal ion requirements of L. major dUTPase, different amounts of MgCl2, MnCl2, CaCl2, CuCl2, and CoCl2 were added to reaction mixtures containing 2500 μM BICINE, 50 μM cresol red, and 1 mg ml−1 of BSA. The reaction was measured at 25°C and pH 8, using the same stopped-flow spectrophotometer.
Inhibition
The inhibition of L. major dUTPase by DMT-dU and α-β-imido-dUTP was determined by including the inhibitor at various concentrations in the assay mixture (50μM cresol red, 2500 μM BICINE, 0.03 μM dUTPase, and 50 μM dUTP). All the inhibition reactions were performed at 25 °C and pH 8. For each inhibitor concentration, the apparent Kmapp value was obtained using the integrated Michaelis–Menten equation as described for dUTP. Km values are a linear function of the inhibitor concentration in competitive systems with product inhibition: 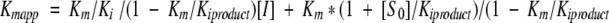 ). A replot of Kmapp versus [I] has intercepts of Km*(1 + [S0]/Kiproduct)/(1 − Km/Kiproduct) on the Kmapp axis. This way the Ki value for the inhibitor can be obtained from the slope value of the linear plot of Kmapp versus [I] and takes into account product inhibition (Segel 1975).
). A replot of Kmapp versus [I] has intercepts of Km*(1 + [S0]/Kiproduct)/(1 − Km/Kiproduct) on the Kmapp axis. This way the Ki value for the inhibitor can be obtained from the slope value of the linear plot of Kmapp versus [I] and takes into account product inhibition (Segel 1975).
The purity of the inhibitor was evaluated on a Mono Q column, using as starting buffer 5 mM PO4H2Na at pH 7 with 50 mM NaCl and 5 mM PO4H2Na with 350 mM NaCl as elution buffer. The flow applied was 1 ml min−1. The hydrolysis of the inhibitor was monitored in the same way but analyzing a mixture of dUTPase and inhibitor after an overnight incubation.
Acknowledgments
We gratefully thank Dr. Nils Gunnar Johansson, Medivir AB, Sweden, for kindly providing the nucleoside triphosphate analog α-β-imido-dUTP. These studies were supported by grants from the Spanish Programa Nacional de Biotecnología (BIO97-0659), the EC BIOMED project contract No.CT97-PL962711 and the Plan Andaluz de Investigación (Cod. CVI-199). V.V-B. is a fellow of the Ramon Areces Foundation. F.H-Z. is a CSIC-Glaxo Wellcome predoctoral fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
dUTPase, dUTP nucleotidohydrolase
α-β-imido-dUTP, 2`-deoxyuridine 5`-(α-β-imido) triphosphate
dUDPase, dUDP nucleotidohydrolase
dNTP, 2`-deoxynucleoside 5`-triphosphate
DMT-dU, 5`-O-(4-4`-dimethoxytrityl)-2`-deoxyuridine
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1101/ps.48801.
References
- Bergman, A.C., Bjornberg, O., Nord, J., Nyman, P.O., and Rosengren, A.M. 1994. The protein p30, encoded at the gap-pro junction of mouse mammary tumor virus, is a dUTPase fused with a nucleocapsid protein. Virology 204 420–424. [DOI] [PubMed] [Google Scholar]
- Bergman, A.C., Bjornberg, O., Nord, J, Rosengren, A.M., and Nyman, P.O. 1995. dUTPase from equine infectious anemia virus: High-level expression in Escherichia coli and purification. Prot. Expr. Purif. 6 379–387. [DOI] [PubMed] [Google Scholar]
- Bernier-Villamor, V., Camacho, A., González-Pacanowska, D., Cederdgen-Zeppezauer, E., Antson, A., and Wilson, K.S. 1999. Crystallisation and preliminary X-ray diffraction of Trypanosoma cruzi dUTPase. Acta Crystallogr. D 55 528–530. [DOI] [PubMed] [Google Scholar]
- Bertani, L.E., Haggmark, A., and Reichhard, P. 1961. Synthesis of deoxyribonucleoside diphosphates with enzymes from Escherichia coli. J. Biol. Chem. 236 PC67–PC68. [PubMed]
- Bjornberg, O. and Nyman, P.O. 1996. The dUTPases from herpes simplex virus type 1 and mouse mammary tumour virus are less specific than the Escherichia coli enzyme. J. Gen. Virol. 773107–3111. [DOI] [PubMed] [Google Scholar]
- Bjornberg, O., Bergman, A.C., Rosengren, A.M., Persson, R., Lehman, I.R., and Nyman, P.O. 1993. dUTPase from herpes simplex virus type 1: Purification from infected green monkey kidney (vero) cells and from an overproducing Escherichia coli strain. Prot. Expr. Purif. 4 149–159. [DOI] [PubMed] [Google Scholar]
- Camacho, A., Arrebola, R., Peña-Diaz, J., Ruiz-Perez, L.M., and González-Pacanowska, D. 1997. Description of a novel eukaryotic deoxyuridine 5`-triphosphate nucleotidohydrolase in Leishmania major. Biochem. J. 325 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, A., Hidalgo-Zarco, F., Bernier-Villamor, V., Ruiz-Perez, L.M., and Gonzalez-Pacanowska, D. 2000. Properties of Leishmania major dUTP nucleotidohydrolase, a distinct nucleotide-hydrolysing enzyme in kinetoplastids. Biochem. J. 346 163–168. [PMC free article] [PubMed] [Google Scholar]
- Cedergen-Zeppezauer, E.S., Larsson, G., Nyman, P.O., Dauter, Z., and Wilson, K.S. 1992. Crystal structure of a dUTPase. Nature 355 740–743. [DOI] [PubMed] [Google Scholar]
- Climie, S., Lutz, T., Radul, J., Summer-Smith, M., Vanderberg, E., and McIntosh, E.M. 1994. Expression of trimeric human dUTP pyrophosphatase in Escherichia coli and purification of the enzyme. Prot. Expr. Purif. 5 252–258. [DOI] [PubMed] [Google Scholar]
- Cornish-Bowden, A. 1995. Introduction to enzyme kinetics. In Fundamentals of enzyme kinetics, 1st ed., pp. 44–47. Portland Press, UK.
- Dauter, Z., Persson, R., Rosengren, A.M., Nyman, P.O., Wilson, K.S., and Cedergren-Zeppezauer, E.S. 1999. Crystal structure of dUTPase from equine infectious anaemia virus; active site metal binding in a substrate analogue complex. J. Mol. Biol. 285 655–673. [DOI] [PubMed] [Google Scholar]
- Dawson, R.M.C., Elliott, D.C., Elliott, W.H., and Jones, K.M. 1986. In Data for biochemical research 3rd edition (ed. ), Oxford University Press, New York.
- El-Hajj, H.H., Zhang, H., and Weiss, B. 1988. Lethality of a dut (deoxyuridine triphosphate) mutation in Escherichia coli. J. Bacteriol. 170 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsden, M.H., McIntosh, E.M., Game, J.C., Wilson, P.J., and Haynes, R.H. 1993. dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. EMBO J. 12 4425–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, I. 1987. "Overproduction, purification and characterization of deoxyuridine-5`-triphosphate nucleotidohydrolase from Escherichia coli." Doctoral thesis, University of Saarbrucken, Germany.
- Kooning, E.V. 1996 Pseudouridine-synthases: Four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res. 24 2411–2415. [DOI] [PMC free article] [PubMed]
- Kornberg, A. and Baker, T.A. 1991. In DNA replication 2nd edition (ed. ), pp. . W.H. Freeman & Co., New York.
- Larsson, G., Svensson, L.A., and Nyman, P.O. 1996a. Crystal structure of the Escherichia coli dUTPase in complex with a substrate analogue (dUDP). Nat. Struct. Biol. 3 32–538. [DOI] [PubMed] [Google Scholar]
- Larsson, G., Nyman, P.O., and Kvassman, J.O. 1996b. Kinetic characterisation of dUTPase from Escherichia coli. J. Biol. Chem. 39 24010–24016. [DOI] [PubMed] [Google Scholar]
- McIntosh, E.M., Looser, J., Haynes, R.H., and Pearlman, R.E. 1994. MluI-site dependent transcriptional regulation of the Candida albicans dUTPase gene. Curr. Genet. 26 415–421. [DOI] [PubMed] [Google Scholar]
- Mol, C.D., Harris, J.M., McIntosh, E.M., and Tainer, J.A. 1996. Human dUTP pyrophosphatase: uracil recognition by a β-hairpin and active sites formed by three separate subunits. Structure 4 1077–1092. [DOI] [PubMed] [Google Scholar]
- Nord, J. 2000. "Nucleotide binding to trimeric dUTPase." Doctoral thesis, University of Lund, Sweden.
- Nord, J., Larsson, G., Kvassman, J.O., Rosengren, A.M., and Nyman, P.O. 1997. dUTPase from the retrovirus equine infectious anemia virus: Specificity, turnover and inhibition. FEBS Lett. 414 271–274. [DOI] [PubMed] [Google Scholar]
- O'Brien, W.E. 1976. A continous spectophotometric assay for argininosuccina synthetase based on pyrophosphate formation. Anal. Biochem. 76 423. [DOI] [PubMed] [Google Scholar]
- Persson, T., Larsson, G., and Nyman, P.O. 1996. Synthesis of 2`-deoxyuridine 5`-(α,β-imido) triphosphate: A substrate analogue and potent inhibitor of dUTPase. Bioorg. Med. Chem. 4 553–556. [DOI] [PubMed] [Google Scholar]
- Prasad, S.G., Stura, E.A., McRee, D.E., Laco, G.S., Hasselkus-Light, C., Elder, J.H., and Stout, C.D. 1996. Crystal structure of dUTP pyrophosphatase from feline immunodeficiency virus. Protein Sci. 5 2429–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad, S.G., Stura, E.A., Elder, J.H., and Stout, C.D. 2000. Structures of feline immunodeficiency virus dUTP pyrophosphatase and its nucleotide complexes in three crystal forms. Acta Crystallogr. D 56 1100–1109. [DOI] [PubMed] [Google Scholar]
- Price, A.R. and Frato, J. 1975. Bacillus subtilis deoxyuridine triphosphate and its bacteriophage PBS2-induced inhibitor. J. Biol. Chem. 250 8804–8811. [PubMed] [Google Scholar]
- Segel, I.H. 1975. Kinetics of unireactant enzymes. In Enzyme kinetics: Behaviour and analysis of rapid equilibrium and steady-state enzyme systems, 1st ed., pp. 54–64. Wiley-Interscience.
- Tye, B.-K., Nyman, P.O., Lehman, I.R., Hochhauser, S., and Weiss, B. 1977. Transient accumulation of Okazaki fragments as a result of uracil incorporation into nascent DNA. Proc. Natl. Acad. Sci. 74 154–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalud, P., Wachs, W.O., Nyman, P.O., and Zeppezauer, M.M. 1994. Inhibition of the proliferation of human cancer cells in vitro by substrate-analogues inhibitors of dUTPase. Adv. Exp. Med. Biol. 370 135–138. [DOI] [PubMed] [Google Scholar]