

Abstract
Biological electron transfer is an efficient process even though the distances between the redox moieties are often quite large. It is therefore of great interest to gain an understanding of the physical basis of the rates and driving forces of these reactions. The structural relaxation of the protein that occurs upon change in redox state gives rise to the reorganizational energy, which is important in the rates and the driving forces of the proteins involved. To determine the structural relaxation in a redox protein, we have developed methods to hold a redox protein in its final oxidation state during crystallization while maintaining the same pH and salt conditions of the crystallization of the protein in its initial oxidation state. Based on 1.5 Å resolution crystal structures and molecular dynamics simulations of oxidized and reduced rubredoxins (Rd) from Clostridium pasteurianum (Cp), the structural rearrangements upon reduction suggest specific mechanisms by which electron transfer reactions of rubredoxin should be facilitated. First, expansion of the [Fe—S] cluster and concomitant contraction of the NH • • • S hydrogen bonds lead to greater electrostatic stabilization of the extra negative charge. Second, a gating mechanism caused by the conformational change of Leucine 41, a nonpolar side chain, allows transient penetration of water molecules, which greatly increases the polarity of the redox site environment and also provides a source of protons. Our method of producing crystals of Cp Rd from a reducing solution leads to a distribution of water molecules not observed in the crystal structure of the reduced Rd from Pyrococcus furiosus. How general this correlation is among redox proteins must be determined in future work. The combination of our high-resolution crystal structures and molecular dynamics simulations provides a molecular picture of the structural rearrangement that occurs upon reduction in Cp rubredoxin.
Keywords: Redox protein, rubredoxin, crystal structure, electron transfer, molecular dynamics
Electron transfer is one of the most essential reactions in life, occurring in processes such as photosynthesis, respiration, and nitrogen fixation. Electron transfer reactions provide the means for transforming solar and chemical energy into a utilizable form in all living organisms. A considerable body of work has elucidated many aspects of the electron transfer process in biological systems (Marcus and Sutin 1985; Moser et al. 1992; Farid et al. 1993; Gray and Ellis 1994; Gray and Winkler 1996). The driving force for transferring electrons from a donor to an acceptor is determined by the magnitude and the sign of the redox potential. Electron transfer proteins can modulate the reduction potential of their redox sites, with differences of up to a few hundred millivolts between homologous proteins with the same redox site and even larger differences between nonhomologous proteins with the same redox site (Cammack 1992). Proteins may regulate electron transfer by gating processes, such as conformational changes (Mei et al. 1999), coupling to ATP hydrolysis (Lanzilotta et al. 1998), and proton transfer (Graige et al. 1998; Regan et al. 1998). However, the basic mechanisms of redox potential control are not fully understood and remain the subject of intense research. Nonheme iron–sulfur [Fe—S] cluster-containing proteins are an important class of redox proteins. They are ubiquitous in living matter and contain sites with one to eight iron atoms (Beinert et al. 1997; Johnson 1998; Bouton 1999). Rubredoxin is the simplest iron–sulfur redox protein and is found in bacteria and archaea (Beinert et al. 1997). It is known to participate in electron transfer reactions in sulfur, nitrate, and superoxide reduction (Seki et al. 1988; Seki et al. 1989; Eggink et al. 1990; Gomes et al. 1997; Jenney et al. 1999). Rubredoxin consists of a short polypeptide chain (52–54 amino acid residues), and its redox site contains a single iron atom, which is tetrahedrally coordinated by four cysteinyl thiolates. The formal redox couples involve Fe(III) and Fe(II); however, the electron density of the cysteinyl sulfurs is known to be equally important (Noodleman et al. 1985). The reduction potentials of rubredoxins range from −60 to +40 mV (Cammack 1992). The relative simplicity and wealth of available data make rubredoxin a powerful model system for investigating the fundamental molecular behavior of proteins. In particular, rubredoxin was one of the first proteins to be analyzed crystallographically at high resolution (Watenpaugh et al. 1973). In reduction potential studies of Clostridium pasteurianum (Cp) rubredoxin (Rd), electrostatic effects do not play a significant role, based on variants with surface charge–change mutations (Zeng and Smith 1996). The net charge of Cp Rd may be increased or decreased, and, in either case, the reduction potential of the Cp Rd variant increases, indicating that simple Coulombic electrostatic effects do not dominate redox potential tuning in Cp Rd. Instead, a structural determinant causing a 50-mV shift in reduction potential between two different groups of rubredoxins has been predicted from computational studies (Swartz et al. 1996) and confirmed experimentally (Eidsness et al. 1999). However, there is some indication that charged side chains may play a role in differentiating reduction potentials in the HiPIPs (Bertini et al. 1997).
Comparisons of global protein structures do not yet allow us to predict reduction potential differences among proteins. Determinants of reduction potentials are believed to come from the local protein structure that surrounds the redox center. To elucidate these structural features, atomic resolution data are needed. We have determined the three-dimensional crystal structures of the oxidized and reduced forms of rubredoxin from C. pasteurianum (Cp Rd) to understand the underlying structural features responsible for its redox reaction. Although other crystal studies of different oxidation states of the same protein have derived one of the states by oxidizing or reducing the already crystallized protein in the opposite state (Takano and Dickerson 1981a, b; Michael et al. 1992; Stout 1993; Schipke et al. 1999), here the protein is in its final oxidation state during the crystallization while maintaining the same pH and salt conditions. Thus, the crystal contacts will not hinder structural changes upon change in oxidation state. Our approach yields significant differences in water molecule movement in the crystal structure of Cp Rd compared to the crystal structure of reduced Pyrococcus furiosus. Significant differences in the water structure have been observed in NMR structures of cytochrome c in the oxidized and reduced state (Qi et al. 1994), which had not been seen in the crystal structures (Takano and Dickerson 1981a, b). However, such differences have not been seen in crystal structures presumably because the change of oxidation state of the protein in the crystalline state may hamper the movement of water into or out of the protein. This study presents the first such evidence from crystallographic experiments, and demonstrates that water motion can be detected when the crystallization is performed for the protein in its final oxidation state. This type of systematic study of high-resolution crystal structures should provide essential data to better understand electron transfer reactions in proteins.
Results
Crystallization
We succeeded in obtaining both reduced and oxidized forms of Cp Rd crystals in the same pH and salt conditions by adjusting the reservoir and crystallization drop conditions. The reduced-state crystals were made by adding a minimum amount of sodium dithionite (Na2S2O4) to the reservoir. Oxidized crystals were made by instead adding an equivalent amount of sodium chloride in order to minimize the vapor pressure difference between the two crystallization processes. The two forms crystallized in the same space group, R3, with one molecule in the asymmetric unit, but they show differences in their unit cell. We have collected 1.5 Å resolution data of both reduced and oxidized forms under a −160°C N2 stream at the Stanford Synchrotron Radiation Laboratory (Beam Line I-5). Although the structures of rubredoxin from P. furiosus (Pf Rd) in its oxidized and reduced state have been reported (Michael et al. 1992), there is a caveat in the crystal structure of the reduced form because the reduced crystals were obtained by adding sodium dithionite directly to the drop-containing crystals of the oxidized form of rubredoxin. It was reported that upon addition of sodium dithionite, the crystals became colorless and extremely fragile (Michael et al. 1992). The crystal was then placed immediately in a −160°C nitrogen stream for the data collection. The structural changes upon reduction were therefore possibly limited through crystal contact forces. Consequently, the unit cell dimensions were virtually identical to those of the oxidized form. Additionally, there may have been differences in pH and/or solvent activity in the two redox states. The overall structures of both forms of Cp Rd are very similar to the other rubredoxins that have been refined to high resolution (Watenpaugh et al. 1980; Sieker et al. 1986; Frey et al. 1987; Adman et al. 1991; Blake et al. 1992; Michael et al. 1992; Dauter et al. 1996; Bau et al. 1998; Misaki et al. 1999). The structures of oxidized forms of Cp Rd from this work and a published work (1IRO [Dauter et al. 1996]) have rms differences of all atoms and only backbone atoms of 0.44 Å and 0.18 Å, respectively. Notable features of typical Rd structures include a three-stranded antiparallel β sheet consisting of the first fourteen residues of the amino terminus and the last five residues of the carboxyl terminus. The remaining structure consists of numerous loops and turns, two of which contain the iron ligands Cys 6, 9, 39, and 42.
Comparison of oxidized and reduced structures of rubredoxin
Changes in the overall crystal structures of the oxidized and reduced forms of Cp were examined and compared with results for the independently determined oxidized (1CAA) (Michael et al. 1992) and reduced (1CAD) (Michael et al. 1992) forms of Pf Rd. These changes indicate the degree of structural relaxation upon reduction of the protein. The rms differences in coordinates upon reduction considering all atoms or only backbone atoms are 0.77 Å and 0.18 Å, respectively, for Cp Rd (excluding the two C-terminal residues out of 54 residues), and are 0.44 Å and 0.20 Å, respectively (Michael et al. 1992), for Pf Rd (excluding the C-terminal residue out of 53 residues). This indicates that the side chains relax more than the backbone, especially in our results for Cp Rd. Also, the radius of gyration (excluding the same residues as in the rms differences) of Cp Rd changes from 9.68 Å to 9.63 Å upon reduction while that of Pf Rd changes from 9.62 Å to 9.61 Å. This indicates that the protein becomes slightly more compact as the redox site becomes more negative, which is reasonable because the protein surrounding the redox site has a positive polarization (Yelle et al. 1995). The changes are small because changes at short distances are less heavily weighted in the radius of gyration. Nevertheless, our results for Cp Rd show slightly greater changes upon reduction than for Pf Rd. Finally, the structural relaxation energies (see Materials and Methods) for only the backbone and for the backbone plus polar side chains are −6 and −11 kcal/mole, respectively, for Cp Rd and −9 and −7 kcal/mole, respectively, for Pf Rd. These results indicate that there is significant relaxation in the protein, but that the polar side chains are actually strained in the Pf Rd results.
Overall, these results indicate that there are significant changes in the structure and energetics in our reduced structure for Cp Rd obtained by crystallizing reduced protein that are not seen in the reduced structure for Pf Rd obtained by reducing the oxidized structure within the constraint of the crystal lattice. In particular, there is significantly more relaxation of the side chains and perhaps less relaxation of the backbone in our results. Thus, reduction in solution may allow more rearrangement of the side chains, whereas reduction in the crystal lattice may force the rearrangement to occur in the backbone to compensate for less overall freedom of movement.
Iron–sulfur cluster environment
Both the oxidized and reduced form of Cp Rd have five hydrogen bonds of NH • • • S type: two to Cys 39 Sγ from Leu 41 NH and Cys 42 NH, two to Cys 6 Sγ from Val 8 NH and Cys 9 NH, and one to Cys 9 Sγ from Tyr 11 NH (Fig. 1a,b ▶). The distances between Cys 42 Sγ and Val 44 NH in both oxidized (3.72 Å) and reduced (3.84 Å) forms of Cp Rd are probably too long for a strong hydrogen bond, unlike the crystal structure of Pf Rd (Michael et al. 1992), in which the valine is replaced by an alanine, which allows the backbone NH group to approach closer to the Cys 42 Sγ. As in other rubredoxins, both Cys 6 and Cys 39 are in the hydrophobic core, but Cys 9 and Cys 42 are facing the surface of the protein.
Fig. 1.

The distances between Fe and Sγ, and between Sγ and amide N for (a) the reduced form of Cp Rd, (b) the oxidized form of Cp Rd. In reduced Cp Rd, the bond lengths between Fe and Sγ are longer by an average of 0.10 ± 0.03 Å and the bond lengths between Sγ and backbone NH are shorter by an average of 0.08 ± 0.05 Å than those of the oxidized form. The distance between Cys 42 Sγ and Val 44 N is not included because this distance is too long to be considered as a regular hydrogen bond.
Reduction of Cp Rd is accompanied by an increase in the four iron–sulfur bond lengths by an average of 0.10 Å (SD = 0.03 Å) consistently in all four Fe—S bonds. It should be noted that these bond lengths were not restrained during final refinement cycles. However, in the previously mentioned study (Michael et al. 1992) of Pf Rd (1CAA, 1CAD), the average Fe—S bond length increases by only 0.04 Å. The degree of change in the Fe—S bond length found here (0.10 Å) is more consistent with a study of the bis(o-xylyl-α,α′-dithiolato)ferrate rubredoxin analog (Lane et al. 1977) and high-potential iron protein (HiPIP) (Carter et al. 1974) in which the average Fe—S bond length increased by 0.09 Å and 0.1 Å, respectively, although the latter structure is at a lower resolution. This expansion of the Fe—S bonds has been viewed in terms of greater antibonding character in the reduced state by the unpaired electrons (Carter et al. 1974).
There is also a decrease in the average NH • • • S hydrogen bond distance by 0.08 Å (SD = 0.05 Å) upon reduction consistently in all five NH • • • S bonds. The contraction of these NH • • • S bonds would help stabilize the negative charge introduced mainly onto the sulfur atoms upon reduction and is consistent with observed shortenings of the corresponding distances upon reduction of Pf Rd (0.09 Å) (Michael et al. 1992) and HiPIP (∼0.1 Å) (Carter et al. 1974).
Leu 41 residue and water penetration
In addition to the structural changes of the Fe—S cluster, there is another structural change that implicates the side chain of Leu 41 in the electron transport mechanism. Leucine 41 is adjacent to the Cys 42 ligand of the redox site and is found at the surface of the protein. In the reduced structure, the side chain of Leu 41 adopts two different conformations, which refine with 60% and 40% relative occupancy (Fig. 2a ▶). However, in the oxidized structures, this side chain clearly has a single conformation (Fig. 2b ▶) similar to the 60% occupancy conformation in the reduced state. We note that in a previous structure of oxidized Cp Rd (1IRO), Leu 41 was found to have two conformations (Dauter et al. 1996); thus, various omit maps in this area were carefully inspected.
Fig. 2.

The electron density map (2F0–Fc) around residue 41 for (a) the reduced form and (b) the oxidized form. In the reduced form, there are two different conformations of Leu 41 side chain. When the side chain of Leu 41 is in the open conformation, a water molecule (depicted by a star) can be placed in the electron density on the other side in hydrogen bonding distance of the Sγ atom of Cys 9.
Interestingly, the two conformations of Leu 41 in the reduced state lead to very different water structure at the redox site. In the 40% occupancy conformation of Leu 41, there is a water molecule in hydrogen bonding distance of Cys 9 Sγ and a distinct ordering of a series of water molecules following it (Fig. 3 ▶). Therefore, one conformation (40% occupancy) of residue 41 in reduced Cp Rd allows the penetration of water molecules, which, in turn, allows the formation of a string of water molecules attached to the Cys 9 Sγ. These results clearly show that the reduced form has increased amounts of hydration around the redox site. It has six water molecules within an 8 Å radius around the Fe center, whereas the oxidized form has only one water molecule within the same radius. Contrary to this observation, no extra water was observed in the reduced crystal structure of Pf Rd. Upon reduction and concomitant increased hydration around the redox site, the backbone of the residues 42 and 43 becomes more rigid as seen in the lowered temperature factors (Fig. 4 ▶). This is most likely caused by the formation of a hydration net around the carbonyl oxygens of residues 42 and 43, which are exposed to the surface. The two conformations also lead to very different electrostatic potential surfaces (Fig. 5 ▶). In the oxidized state, Sγ of Cys 42, which does not have any hydrogen bonds to it from any backbone amides, is the most solvent exposed atom of the Fe—S cluster. On the other hand, in the reduced state, the opening of the Leu 41 gate leads to an extra electronegative potential hole around the Cys 9 Sγ on the surface of the protein.
Fig. 3.

Local structure of the reduced form around Fe—S cluster and Leu 41 in its open conformation (open water gate). Placing the side chain of Leu 41 away from Cys 9 Sγ allows the string of water molecules to approach the Cys 9 Sγ, with the first water forming a hydrogen bond with Cys 9 Sγ.
Fig. 4.

The backbone temperature factor plot of the oxidized and reduced forms. Values of the oxidized form are marked as closed diamonds connected with a dotted line and those of the reduced form as closed squares connected with a solid line. In the reduced form structure, Leu 41, Cys 42, and Gly 43 have lower temperature factors compared to those of the oxidized form.
Fig. 5.

The electrostatic potential surfaces of (a) the oxidized form and (b) the reduced form (in open water gate state). Upon reduction and consequent opening of the gate, Cys 9 Sγ exposes its electronegative potential surface to the solvent, and this extra electronegative potential hole could attract the string of water molecules.
Earlier molecular dynamics studies of Pf (Swartz and Ichiye 1996) and Cp (Yelle et al. 1995) rubredoxins have revealed that water is able to easily penetrate into the redox site area via nonpolar side chain movement in the reduced state, but is not able to penetrate in the oxidized state on the time scale of the simulations. Thus, the movements of the nonpolar side chains have been proposed to act as gates controlling access of water to the redox site, with greater solvent penetration in the reduced state owing to the higher net charge of the redox site cluster (the couple is Fe(III)(SR)4− and Fe(II)(SR)42−) (Yelle et al. 1995). Given these earlier results, new, longer simulations of Cp rubredoxin are presented here, which also show water entry in the reduced state but not the oxidized state. In the simulation of the reduced form, the Leu 41 gate remains open with two waters entering and exiting from positions 3.2 Å from Cys 9 Sγ. The first water, which is in the crystal structure position, has occupancy of ∼65%, with a residence time of ∼4 psec and absence time of ∼3 psec. The second water, located ∼4 Å away from the first water, has an occupancy of ∼65%, with a residence time of ∼10 psec and absence time of ∼4 psec. The gate apparently tries to close occasionally during the reduced simulation; however, water is still present in the crystal structure position and sometimes the second position during these times so that the gate cannot fully close. In the simulations of the oxidized form, the Leu 41 gate opens and closes, but no waters enter. Also, in the simulation of the reduced form, a third water enters to reside near Cys 42 Sγ (3.53 Å). It appears to be controlled by a breathing motion of the Val 8 and Val 44 side chains and has occupancy of ∼90%. This water is not seen in the simulation of the oxidized form. However, both forms of crystal structures do not show any water molecule closer than 5.0 Å from the Cys 42 Sγ.
The simulations only cover periods when the gate is closed or open but with no water present for the oxidized state and when the gate is open with water present for the reduced state. However, our interpretation is that in the real protein in solution, the Leu 41 gate opens and closes in the reduced state as seen in our crystal structure, and in the oxidized state because the open state has been observed in other crystal structures of oxidized Cp Rd (1IRO [Dauter et al. 1996]). For reduced rubredoxin in solution, the water can enter and exit when the gate is open, attracted by the stronger electronegative potential hole. The gate is prevented from closing if a water is present but does not necessarily shut if the water is not present. When the gate is completely closed in the reduced state, which we have not seen in simulation, presumably no water is present. However, for oxidized rubredoxin in solution, if the gate is open, the electronegative hole will not be as strong because of the lower electron density on the sulfurs in the oxidized relative to reduced state, so that water rarely enters. Thus, the gate is likely to be closed more often in the oxidized state because no water molecules are present to prevent the gate from closing.
It has been long recognized that reduction potentials depend on the proximity of the redox active site and solvent (Stephens et al. 1996). The difference in the water structure near the redox site in the oxidized versus reduced states will obviously affect the reduction potential. The presence of a polar water molecule near the redox site in the reduced state will greatly enhance the affinity of the site for an electron by making the electrostatic environment more favorable, thus increasing the reduction potential over the case where a water molecule is not present. In fact, calculations indicate that the three water molecules in the molecular dynamics simulations of Cp Rd will contribute ∼800 mV to the electrostatic potential and that the single water in the crystal structures of Cp Rd will contribute ∼240 mV to the electrostatic potential assuming 40% occupancy.
Discussion
One of the most intriguing questions about redox proteins is how the protein environment influences the electron transfer properties of its redox site and what underlying structural features are responsible for the redox reaction. Based on the observed information with two 1.5 Å resolution structures, we have come to the following picture of how the redox reaction is controlled in rubredoxin. The concerted structural rearrangements that occur upon reduction suggest specific mechanisms by which electron transfer reactions are facilitated: (1) The expansion of the [Fe—S] cluster and contraction of the NH • • • S bonds stabilizes the extra negative charge, which is mainly on the sulfurs, and perhaps leads to electrostatic buffering by the NH • • • S bonds so that structural perturbations of the rest of the protein are minimized. (2) A gating mechanism arising from the conformational change of Leu 41, a nonpolar side chain, allows a transient penetration of water molecules, providing an electrostatic charge–dipole interaction between the negatively charged cluster and the string of water molecules, and possibly also providing a source of protons.
The water gating by a hydrophobic side chain presented here is a mechanism that can promote the reduction of the Fe—S cluster. Moreover, it is tempting to speculate on how the water gating can promote electron transfer in an electron transport chain. First, an electron would be transferred from an electron donor protein to an oxidized rubredoxin, which would result in a reduced rubredoxin. This reduced rubredoxin could be stabilized by the transient entry of water through the gate. Next, the stabilized reduced rubredoxin could diffuse to a partner electron-acceptor protein. On the diffusion time scale, the gate could open and close many times. If acceptor protein bound while the gate is closed and no water is present in a manner that hinders the gate from opening, the reduced rubredoxin would be kept in the less stable state without the water. This would favor electron transfer to the acceptor protein, which could occur through the exposed Cys 42 Sγ. Alternatively, the water in the reduced rubredoxin could provide its proton to a partner acceptor protein such as superoxide reductase (SOR) in P. furiosus (Jenney et al. 1999). After the proton donation, the reduced rubredoxin would again be in the less stable state, thus favoring subsequent electron transfer to the same partner acceptor protein. A similar mechanism has been recently described for the ferredoxin (Fd I) from Azotobacter vinelandii in which the side chain of a surface Asp residue plays a critical role in proton coupling and gating in the reduction and oxidation cycle (Chen et al. 2000).
In summary, the availability of high resolution structures of Cp rubredoxin crystallized in the oxidized and reduced states under the same crystallization conditions allows us to identify differences upon reduction, which may be obscured by the crystal contacts when an oxidized crystal is reduced. These differences are crucial in understanding the structural basis of reduction potentials of electron transfer proteins.
Materials and methods
Overexpression and purification of Cp wild-type rubredoxin
Escherichia coli strain BL21-DE3 containing plasmid of Cp Rd wild type was grown while shaking (250 rpm) at 37°C in 4 L of Luria–Bertani medium supplemented with ampicillin (100 μg/mL), until the optical density at 600 nm reached 1.2–1.5, at which time 0.4 mM isopropyl β-d-thiogalactoside was added. The culture was grown further for 8–10 h and harvested by centrifugation for 20 min at 6000g. The cell pellet was resuspended in 50 mL of 50 mM Tris-HCl buffer at pH 8.0. The cells were then lysed by sonication (Tekmar Sonic Disruptor), and the lysate was cleared by centrifugation at 27,000g for 2 h. Cell lysate was applied onto a DEAE column equilibrated with 50 mM Tris-HCl buffer at pH 8.0. Cp Rd was then eluted with a gradient application of the elution buffer containing 1M NaCl. The eluted Cp Rd was desalted and concentrated by ultrafiltration in an Amicon cell with a 3K MWCO membrane, YM3 (Amicon) in 50 mM Tris-HCl at pH 8.5 and applied to a Uno Q-12 column (Bio-Rad) connected to a BioCAD perfusion chromatography system (Perceptive Biosystem Inc.). The final purification was performed using a hydroxyapatite column (CHT10-I, Bio-Rad) with an equilibrating buffer of 5mM sodium phosphate at pH 6.0 and an eluting buffer of 500 mM sodium phosphate at pH 6.0. The Cp Rd wild-type protein, which eluted at 14%–20% of the eluting buffer, was collected and concentrated to 30 mg/mL after changing the buffer with 0.1 M sodium acetate (pH 4.6) for crystallization. The Cp Rd protein was characterized by its UV-visible absorption spectra and electrospray ionization mass spectrometry.
Crystallization of oxidized and reduced forms of Cp rubredoxin
The crystals of the oxidized Cp Rd were grown by the vapor diffusion method in 2.0 M ammonium sulfate and 0.1 M sodium acetate at pH 4.6. In addition, the reduced form crystals had 0.1 g of sodium dithionite added in the 0.5 mL of reservoir solution, and the oxidized form had an equivalent amount of NaCl instead. For hanging drop crystallization, 2 μL of this reservoir solution containing the reducing reagent was added to the 2-μL drop of protein solution. The procedures for the reduced form were performed under a gaseous nitrogen stream. Deep-red-colored crystals of the oxidized form and transparent, colorless crystals of the reduced form were grown in 24 h to the size of 0.3 × 0.3 × 0.3 mm. X-Ray diffraction analysis revealed that both forms of Cp Rd were crystallized in a trigonal space group R3. The unit-cell parameters of oxidized Cp Rd were a = b = 64.115, c = 32.183 Å, and those of reduced Cp Rd were a = b = 63.335, c = 32.420 Å.
Data collection, molecular replacement, and crystallographic refinement
Both data sets were collected at the Stanford Linear Accelerator Center (Beam line 1–5) under a −160°C liquid nitrogen stream with a crystal-to-detector distance of 170 mm. To obtain the high-resolution data, 2θ was set to 15°. The final Rsym value of the Cp oxidized form was 5.9%, generating 9328 reflections, and the final Rsym of the Cp reduced form was 4.5%, generating 9224 reflections above the 1σ level. A previous crystal structure of the Cp oxidized form (1IRO [Dauter et al 1996]) was used in solving the structures with the refinement computer program X-PLOR (Brunger 1992). Initial rigid-body refinement was carried out using 1675 reflections for the oxidized and 1602 reflections for the reduced form from 15.0 Å to 2.5 Å resolution data and produced an R value of 22% for the oxidized and 23.6% for the reduced form, respectively. After several cycles of positional refinement, temperature factor refinement, and simulated annealing omit map calculation, we were able to fit all residues to the electron density. The residue 54 (Glu) is disordered in our oxidized structure, and the corresponding electron density was not visible from the early stage of refinement. The R factors for the final models containing 477 nonhydrogen atoms for the oxidized and 502 for the reduced forms are 19.1% and 18.0%, respectively. The reflection numbers above 2σ level were 7630 (99.7% completeness) for the oxidized and 7589 (97.3% completeness) for the reduced forms between 10.0 Å and 1.5 Å resolution. The rms deviations (from standard geometry) of the oxidized form are 0.03 Å for bonds and 3.57° for angles, and those of the reduced form are 0.02 Å and 3.01°, respectively. There are 64 water molecules in the oxidized structure and 79 water molecules in the reduced structure. The coordinates have been deposited in the Brookhaven Protein Data Bank (Bernstein et al. 1977) (1FHH for the oxidized and 1FHM for the reduced).
Molecular dynamics simulations
Molecular dynamics simulations were performed with the molecular mechanics/dynamics program CHARMM (Brooks et al. 1983) using truncated rectangular–octahedral periodic boundary conditions in the microcanonical ensemble at ∼300 K. The Verlet algorithm was used with a time step of 0.001 psec. The potential energy parameters were CHARMM 19 set (Brooks et al. 1983) with implicit nonpolar hydrogens and TIP3P water (Jorgensen 1981) plus additional parameters for the ions and the iron–sulfur site (Yelle 1996). Long-range forces were switched smoothly to zero using an atom-based force-switch method (Steinbach and Brooks 1994) between 10 Å and 14 Å. The starting structure was the 5RXN crystal structure of Cp Rd plus crystal waters. Additional water from a preequilibrated box and counter-ions were added so that the final system consisted of 501 protein atoms, 1835 water molecules, 15 sodium ions, and 5 chlorine ions. The solvent environment was equilibrated with 70 psec of dynamics in which only the water and counter-ions were allowed to move, and then the entire system was equilibrated by more than 10 psec of dynamics. The analysis was performed 400 psec after equilibration.
Energy calculations
The contribution of the waters to the change in electrostatic energy was calculated by
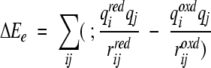 |
in which i is summed over the iron and Cβ and Sγ of the four cysteinyl ligands of the redox site, j is summed over the oxygen and hydrogens of the water, qi is the charge of atom i, rij is the distance from atom i to atom j, and the superscripts oxd and red denote oxidized and reduced, respectively. In the crystal structures, this was calculated explicitly for each water molecule, whereas in the molecular dynamics simulations, this was calculated by integrating the potential multiplied by the radial distribution function from the simulation to a value corresponding to the number of water molecules. The structural relaxation energy was calculated by a similar equation:
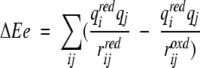 |
in which now j is summed over the specified set of protein atoms and the partial charges are for the reduced state only. No cutoffs or switches in the long-range forces were used for these calculations.
Coordinates
The coordinates have been deposited in the Brookhaven Protein Data Bank (1FHH and 1FHM).
Acknowledgments
We thank J.T. Fisher and H. Bellamy. This work was supported by grants from the National Institutes of Health.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.34501.
References
- Adman, E.T., Sieker, L.C., and Jensen, L. 1991. Structure of rubredoxin from Desulfovibrio vulgaris at 1.5 Å resolution. J. Mol. Biol. 217 337–352. [DOI] [PubMed] [Google Scholar]
- Bau, R., Rees, D.C., Kurtz, D.M. Scott, Jr., R.A., Huang, H., Adams, M.W.W., and Eidsness, M.K. 1998. Crystal structure of rubredoxin from Pyrococcus furiosus at 0.95 Å resolution, and the structures of N-terminal methionine and formylmethionine variants of Pf Rd. Contributions of N-terminal interactions to thermostability. J. Biol. Inorg. Chem. 3 484–493. [Google Scholar]
- Beinert, H., Holm, R.H., and Munck, E. 1997. Iron–sulfur clusters: Nature's modular, multipurpose structures. Science 277 653–659. [DOI] [PubMed] [Google Scholar]
- Bernstein, F.C., Koetzle, T.F., Williams, G.J., Meyer, Jr., E.E., Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T., and Tasumi, M. 1977. The protein data bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 112 535–542. [DOI] [PubMed] [Google Scholar]
- Bertini, I., Gori-Savellini, G., and Luchinat, C. 1997. Are unit charges always negligible? J. Biol. Inorg. Chem. 2 114–118. [Google Scholar]
- Blake, P.R., Park, J.B., Zhou, Z.H., Hare, D.R., Adams, M.W., and Summers, M.F. 1992. Solution-state structure by NMR of zinc-substituted rubredoxin from the marine hyperthermophilic archaebacterium Pyrococcus furiosus. Protein Sci. 1 1508–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouton, C. 1999. Nitrosative and oxidative modulation of iron regulatory proteins. Cell. Mol. Life. Sci. 55 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., and Karplus, M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4 187–217. [Google Scholar]
- Brunger, A.T. 1992. X-PLOR: A system for crystallography and NMR (version 3.1). Yale University, New Haven, CT.
- Cammack, R. 1992. Iron–sulfur cluster in enzymes: Themes and variations. Adv. Inorg. Chem. 38 281–322. [Google Scholar]
- Carter, Jr., C.W., Kraut, J., Freer, S.R., and Alden, R.A 1974. Comparision of oxidation–reduction site geometries in oxidized and reduced Chromatium high potential iron protein. J. Biol. Chem. 249 6339–6346. [PubMed] [Google Scholar]
- Chen, K., Hirst, J., Camba, R., Bonagura, C.A., Stout, C.D., Burgess, B.K., and Armstrong, F.A. 2000. Atomically defined mechanism for proton transfer to a buried redox centre in a protein. Nature 405 814–817. [DOI] [PubMed] [Google Scholar]
- Dauter, Z., Wilson, K.S., Sieker, L.C., Moulis, J.M., and Meyer, J. 1996. Zinc- and iron-rubredoxins from Clostridium pasteurianum at atomic resolution: The first high-precision model of a ZnS4 unit in a protein. Proc. Natl. Acad. Sci. USA 93 8836–8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggink, G., Engel, H., Vriend, G., Terpstra, P., and Witholt, B. 1990. Rubredoxin reductase of Pseudomonas oleovorans. Structural relationship to other flavoprotein oxidoreductases based on one NAD and two FAD fingerprints. J. Mol. Biol. 212 135–142. [DOI] [PubMed] [Google Scholar]
- Eidsness, M.K., Burden, A.E., Richie, K.A., Kurtz, Jr., D.M., Scott, R.A., Smith, E.T., Ichiye, T., Beard, B., Min, T., and Kang, C. 1999. Modulation of the redox potential of the [Fe(SCys)4] site in rubredoxin by the orientation of a peptide dipole. Biochemistry 38 14803–14809. [DOI] [PubMed] [Google Scholar]
- Farid, R.S., Moser C.C., and Dutton, P.L. 1993. Electron transfer in proteins. Curr. Op. Struc. Biol. 3 225–233. [Google Scholar]
- Frey, M.W., Sieker, L., Payan, F., Haser, R., Bruschi, M., Pepe, G., and LeGall, J. 1987. Rubredoxin from Desulfovibrio gigas. A molecular model of the oxidized form at 1.4 Å resolution. J. Mol. Biol. 197 525–541. [DOI] [PubMed] [Google Scholar]
- Gomes, C.M., Silva, G., Oliveira, S., LeGall, J., Liu, M.Y., Xavier, A.V., Rodrigues-Pousada, C., and Teixeira, M. 1997. Studies on the redox centers of the terminal oxidase from Desulfovibrio gigas and evidence for its interaction with rubredoxin. J. Biol. Chem. 272 22502–22508. [DOI] [PubMed] [Google Scholar]
- Graige, M., Feher, G., and Okamura, M.Y. 1998. Conformation gating of the electron transfer reaction QA−•QB → QAQB−• in bacterial centers of Rhodobacter sphaeroides determined by a driving force assay. Proc. Natl. Acad. Sci. USA 95 11679–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray, H.B. and Ellis, Jr., W.R. 1994. Electron transfer. In Bioinorganic chemistry (eds. I. Bertini et al.), pp. 315–363. University Science Books, Sausilito, CA.
- Gray, H.B. and Winkler, J.R. 1996. Electron transfer in proteins. Ann. Rev. Biochem. 65 537–561. [DOI] [PubMed] [Google Scholar]
- Jenney, F.E., Verhagen, M.F.J.M., and Adams, M.W.W. 1999. Anaerobic Microbes: Oxygen detoxification without superoxide dismutase. Science 286 306–309. [DOI] [PubMed] [Google Scholar]
- Johnson, M.K. 1998. Iron–sulfur proteins: New roles for old clusters. Curr. Opin. Chem. Biol. 2 173–181. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W.L. 1981. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Amer. Chem. Soc. 103 335–340. [Google Scholar]
- Lane, R.W., Ibers, J.A., Frankel, R.B., Papaefthymiou, G.C., and Holm, R.H. 1977. Synthetic analogues of the active sites of iron–sulfur proteins. 14. Synthesis, properties, and structures of bis(o-xylyl-a,a′-dithiolato) ferrate(II,III) anions, analogues of oxidized and reduced rubredoxin sites. J. Am. Chem. Soc. 99 84–98. [DOI] [PubMed] [Google Scholar]
- Lanzilotta, W., Parker, V., and Seefeldt, L.C. 1998. Electron transfer in nitrogenase analyzed by Marcus theory: Evidence for gating by MgATP. Biochemistry 37 399–407. [DOI] [PubMed] [Google Scholar]
- Marcus, R.A. and Sutin, N. 1985. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 811 265–322. [Google Scholar]
- Mei, H., Wang, K., Peffer, N., Weatherly, G., Cohen, D.S., Miller, M., Pielak, G., Durham, B., and Millett, F. 1999. Role of configurational gating in intracomplex electron transfer from cytochrome c to the radical cation in cytochrome c peroxidase. Biochemistry 38 6846–6854. [DOI] [PubMed] [Google Scholar]
- Michael, W., Day, M.W., Hsu, B.T., Joshua-Tor, L., Park, J-B., Zhou, Z.H., Adams, M.W.W., and Rees, D.C. 1992. X-Ray crystal structures of the oxidized and reduced forms of the rubredoxin from the marine hyperthermophilic archaebacterium Pyrococcus furiosus. Protein Sci. 1 1494–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaki, S., Morimoto, Y., Ogata, M., Yagi, T., Higuchi, Y., and Yasuoka, N. 1999. Structure determination of rubredoxin from Desulfovibrio vulgaris Miyazaki F in two crystal forms. Acta Cryst. D 55 408–413. [DOI] [PubMed] [Google Scholar]
- Moser, C.C., Keske, J.M., Warncke, K., Farid, R.S., and Dutton, P.L. 1992. Nature of biological electron transfer. Nature 355 796–802. [DOI] [PubMed] [Google Scholar]
- Noodleman, L., Norman, Jr., J.G., Osborne, J.H., Aizman, A., and Case, D.A. 1985. Models for ferredoxins: Electronic structures of iron–sulfur clusters with one, two, and four iron atoms. J. Am. Chem. Soc. 107 3418–3426. [Google Scholar]
- Qi, P.X., Urbauer, J.L., Fuentes, E.J., Leopold, M.F., and Wand, J.A. 1994. Structural water in oxidized and reduced horse cytochrome c. Nature Struct. Biol. 1 378–382. [DOI] [PubMed] [Google Scholar]
- Regan, J., Ramirez, B., Winkler, J.R., Gray, H.B., and Malmstrom, B.G. 1998. Pathways for electron tunneling in cytochrome c oxidase. J. Bioenerg. Biomembr. 30 35–39. [DOI] [PubMed] [Google Scholar]
- Schipke, C.G., Goodin, D.B., McRee, D.E., and Stout, C.D. 1999. Oxidized and reduced Azotobacter vinelandii ferredoxin I at 1.4 Å resolution: Conformational change of surface residues without significant change in the [3Fe—4S]+/0 cluster. Biochemistry 38 8228–8239. [DOI] [PubMed] [Google Scholar]
- Seki, S., Ikeda, A., and Ishimoto, M. 1988. Rubredoxin as an intermediary electron carrier for nitrate reduction by NAD(P)H in Clostridium perfringens. J. Biochem. (Tokyo) 103 583–584. [DOI] [PubMed] [Google Scholar]
- Seki, Y., Seki, S., Satch, M., Ikeda, A., and Ishimoto, M. 1989. Rubredoxin from Clostridium perfringens: Complete amino acid sequence and participation in nitrate reduction. J. Biochem. (Tokyo) 106 336–341. [DOI] [PubMed] [Google Scholar]
- Sieker, L.C., Stenkamp, R.E., Jensen, L.H., Pickril, B., and LeGall, J. 1986. Structure of rubredoxin from the bacterium Desulfovibrio desulfuricans. FEBS Lett. 208 73–76. [DOI] [PubMed] [Google Scholar]
- Steinbach, P.J. and Brooks, B. R. 1994. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comp. Chem. 15 667–683. [Google Scholar]
- Stephens, P.J., Jollie, D.R., and Warshel, A. 1996. Protein control of redox potentials of iron–sulfur proteins. Chem. Rev. 96 2491–2513. [DOI] [PubMed] [Google Scholar]
- Stout, C.D. 1993. Crystal structures of oxidized and reduced Azotobacter vinelandii ferredoxin at pH 8 and 6. J. Biol. Chem. 268 25920–25927. [PubMed] [Google Scholar]
- Swartz, P.D. and Ichiye, T. 1996. Temperature dependence of the redox potential of rubredoxin from Pyrococcus furiosus: A molecular dynamics study. Biochemistry 35 13772–13779. [DOI] [PubMed] [Google Scholar]
- Swartz, P.D., Beck, B.W., and Ichiye, T. 1996. Structural origins of redox potential in iron–sulfur proteins: Electrostatic potentials of crystal structures. Biophys. J. 71 2958–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, T. and Dickerson, R.E. 1981a. Conformational change of cytochrome c. I. Ferrocytochrome c structure refined at 1.5 Å resolution. J. Mol. Biol. 153 79–94. [DOI] [PubMed] [Google Scholar]
- Takano, T. and Dickerson, R.E. 1981b. Conformational change of cytochrome c. II. Ferricytochrome c refinement at 1.8 Å and comparison with the ferrocytochrome structure. J. Mol. Biol. 153 95–115. [DOI] [PubMed] [Google Scholar]
- Watenpaugh, K.D., Sieker, L.C., Herriott, J.R., and Jensen, L.H. 1973. Refinement of the model of a protein: Rubredoxin at 1.5 Å resolution. Acta Cryst. B 29 943–956. [Google Scholar]
- Watenpaugh, K., Sieker, L.C., and Jensen, L.H. 1980. Crystallographic refinement of rubredoxin at 1.2 Å resolution. J. Mol. Biol. 138 615–633. [DOI] [PubMed] [Google Scholar]
- Yelle, R.B. 1996. "Theoretical studies of the electron transfer properties of rubredoxin." Ph.D. thesis, Washington State University, Pullman.
- Yelle, R.B., Park, N.-S., and Ichiye, T. 1995. Molecular dynamics simulations of rubredoxin from Clostridium pasteurianum: Changes in structure and electrostatic potential during redox reactions. Proteins 22 154–167. [DOI] [PubMed] [Google Scholar]
- Zeng, Q. and Smith, E. 1996. Protein determinants of metal site reduction potentials: Site-directed mutagenesis studies of Clostridium pasteurianum rubredoxin. Inorg. Chim. Acta 242 245–251. [Google Scholar]