

Abstract
Streptavidin provides an effective receptor for biotinylated tumoricidal molecules, including radionuclides, when conjugated to an antitumor antibody and administered systemically. Ideally, one would like to administer this bacterial protein to patients repeatedly, so as to maximize the antitumor effect without eliciting an immune response. Therefore, we attempted to reduce the antigenicity of streptavidin by mutating surface residues capable of forming high energy ionic or hydrophobic interactions. A crystallographic image of streptavidin was examined to identify residues with solvent-exposed side chains and residues critical to streptavidin's structure or function, and to define loops. Mutations were incorporated cumulatively into the protein sequence. Mutants were screened for tetramer formation, biotin dissociation, and reduced immunoreactivity with pooled patient sera. Patient antisera recognized one minor continuous epitope with binding locus at residue E101 and one major discontinuous epitope involving amino acid residues E51 and Y83. Mutation of residues E51, Y83, R53, and E116 reduced reactivity with patient sera to <10% that of streptavidin, but these mutations were no less antigenic in rabbits. Mutant 37, with 10 amino acid substitutions, was only 20% as antigenic as streptavidin. Rabbits immunized with either streptavidin or mutant 37 failed to recognize the alternative antigen. Biotin dissociated from mutant 37 four to five times faster than from streptavidin. Residues were identified with previously undescribed impact on biotin binding and protein folding. Thus, substitution of charged, aromatic, or large hydrophobic residues on the surface of streptavidin with smaller neutral residues reduced the molecule's ability to elicit an immune response in rabbits.
Keywords: Streptavidin, variants, antigenicity, site-directed mutagenesis
The immune response is a potential impediment to the repeated or sustained systemic use of any exogenous protein or other macromolecule in humans. Clinical and preclinical therapy studies have shown that exogenous proteins can be effective in vivo as artificial receptors for the capture of radionuclides (Goodwin et al. 1986; Axworthy et al. 2000), as toxins (Wawrzynczak 1991) or as catalysts for the activation of prodrugs (Melton et al. 1990; Senter 1990; Meyer et al. 1993 ). In some cases a human protein can carry out these functions, but frequently human proteins are unavailable or inadequate, or non-human proteins are preferable. The immune response to murine antibodies, for instance, has stimulated a major ongoing field of research aimed at "humanization", that is, converting nonbinding regions of monoclonal antibodies from murine to human sequence (Tamura et al. 2000 and references therein). Except at very high doses, systemic administration of an exogenous protein in a host that has already mounted an immune response produces a substantially altered pharmacokinetic pattern with reduced bioavailability, making dosing uncertain and efficacy unreliable. Consequently, it is important to reduce the immune response sufficiently to preserve "normal" pharmacokinetic behavior.
One approach to reduce the host response to an exogenous protein is to suppress the activity of the immune system. For patients with cancer or other diseases, however, suppressing the immune system may have deleterious effects. Alternatively, the immunogenicity of proteins can be modulated by synthetic modification with polyethylene glycol or dextran (Fagnani et al. 1990; Delgado et al. 1992; Marshall et al. 1996; Mikolajczyk et al. 1996), but these modifications can impair the activity of the protein and by their heterogeneous nature detract from the quality of the product while adding expense. Proteins can also be modified in a manner that induces tolerance in the patient. Zamdidis and Scott (1996) fused a 15-amino-acid peptide containing B-cell and T-cell epitopes of the bacteriophage λ cI repressor protein to the amino terminus of the mouse IgG1 heavy chain. The peptide-grafted IgG1 was tolerogenic in mice for both epitopes. However, the induction of tolerance occurs over time and is not generally practical for patients awaiting therapy. The goal of this study was to develop a mutant form of the exogenous protein streptavidin that retained the essential activity of the protein but with a substantially reduced ability to induce an immune response in animals.
Streptavidin has been used effectively as an artificial receptor for the capture of systemically administered therapeutic radionuclides in preclinical (Axworthy et al. 2000) and human clinical studies (Breitz et al. 2000; Knox et al. 2000). In the Pretarget delivery system, streptavidin, either chemically conjugated or genetically fused to a single chain antibody fragment (Schultz et al. 2000), accumulates at sites recognized by the antibody. An appropriately designed clearing agent removes residual streptavidin from the circulation through the liver's Ashwell receptors. A biotin derivative containing a therapeutic radionuclide is then administered systemically, whereupon it is either captured by streptavidin at the tumor site, or is rapidly excreted through the kidney. In early clinical evaluation of this delivery system, patients with a variety of adenocarcinomas received between 200 and 600 mg of a murine NR-LU-10 monoclonal antibody (mAb)/streptavidin conjugate. ELISA analysis of pooled serum from these patients showed that they mounted an immune response within 10–14 days after treatment and that this response was primarily directed toward the streptavidin portion of the conjugate (Breitz et al. 2000).
We adopted a site-directed mutagenic approach to modifying streptavidin because of the availability of high quality crystallographic data and the absence of a suitable library screening method. The X-ray crystal structure was examined to identify interactive residues (i.e., those that might contribute to antibody recognition), and to avoid mutating residues obviously involved in biotin binding, subunit interaction, or folding and structure stabilization. Surface aromatic residues forming nonbonded contacts with antibody residues could give rise to high affinity binding (Davis and Teague 1999) and therefore were candidates for mutagenesis. Charged residues were also prime candidates for mutagenesis. Although the enthalpy changes associated with interactions between charged or polar residues are small when the loss of hydrogen bonding to solvent is taken into account, they have a major impact on the specificity of the interaction, and therefore are critical to antibody–antigen interaction. It was assumed that amino acids that are small, neutral, and hydrophilic are unlikely to contribute to the antibody response. Thus Ser, Thr, Ala, and Gly residues were generally preserved. Lysine residues were also preserved throughout most of the study because of their utility in conjugation of streptavidin to antitumor antibodies. Because the aim of the study was to facilitate the use of streptavidin in vivo, we focused on residues with solvent-exposed side chains, reasoning that antibody recognition of interior sections of the folded protein would not impact its behavior in circulation.
Mutants were prepared and screened based on four criteria: (1) high yield of properly folded tetramer, (2) reduced recognition by either murine monoclonal anti-streptavidin (early screens), or rabbit anti-mutant 19 (later screens), (3) reduced recognition by human anti-streptavidin from patients, and (4) biotin dissociation rate. Mutants selected from each screen showed the best combination of these four properties.
Although diminished reactivity of a mutant with antisera to the native protein does not guarantee that the mutant is less antigenic, we hypothesized that the physical forces that stabilize antigen–antibody interactions could also be important in antigen recognition by the immune system. Therefore, it seemed reasonable that antigenicity could be reduced if the residues responsible for the preponderance of the binding energy were replaced. In addition, screening for immunoreactivity can be quickly and economically accomplished using ELISA assays, whereas screening for antigenicity cannot. Streptavidin is a homotetramer, each subunit of which contains ∼124 amino acids. Because the 159-amino-acid genomic protein is clipped at both termini by enzymes in the expressing organism Streptomyces avidinii (Pahler et al. 1987), the exact molecular mass can vary and microheterogeneity is observed depending on production means. The streptavidin we studied consists of residues 13–136 of genomic streptavidin ("core streptavidin") but with the amino terminal alanine, A13, replaced with the translation-initiating Met. X-ray crystallography shows that the protein folds in a β-barrel with eight β-strands, in which the surface formed by the amino terminal section (residues ∼13–57) is exposed to the solvent. The remainder forms a large buried hydrophobic sheet that interacts with an adjoining subunit (Hendrickson et al. 1989; Weber et al. 1992a, b). The loops between the β-strands in the buried section of the surface are also exposed to the solvent. The dimers formed by the hydrophobic β-sheet interface are linked into a tetramer by a small hydrophobic region (T123–V125) and His–His π–π interactions (H127) in the carboxy-terminal region of the sequence. Streptavidin is a stable protein, as is evident from its high melting temperature (84.1°C), propensity to crystallize, resistance to moderate denaturing conditions, and propensity to refold (Sano and Cantor 1990a, b, Sano and Cantor 1995; Kurzban 1991; Weber et al. 1994; Sano et al. 1995; Frietag et al. 1997; Katz 1997). We have also observed that core streptavidin is relatively resistant to enzymatic proteolysis.
Streptavidin's propensity to refold (Sano and Cantor 1990b) permits the facile expression of numerous mutants without the need to select optimal expression conditions for each. Its stability allows the incorporation of numerous mutations and provides a clear identification of nonpermissible mutations. Streptavidin's extremely high affinity for biotin (KD ∼10−15 M) provides a wide range over which the impact of mutation on biological function can be measured. Thus, in addition to its use in pretargeting, streptavidin exhibits several properties that make it a useful model with which to test epitope mutation as a means of reducing a protein's antigenicity.
Results
Streptavidin structure
The streptavidin monomer was divided into segments surrounding the loop regions (Loops 1–7 (in capital letters, starting at N terminus); Figs. 1 and 2A ▶ ▶) using the computer software Iditis (Oxford Molecular Ltd., Oxford, UK) that is based on an extension to the Kabsch and Sander classification of secondary structure (Kabsch and Sander 1983). Analysis of the structure showed that the loops themselves account for ∼60% of the solvent-accessible surface. When three amino acids on either side of the loop residues are included in the analysis, 80% of the solvent-accessible surface is accounted for. The β-strand residues on either side of the loops were included in our analyses as they may form hydrogen bonds needed to stabilize the peptides in a turn conformation. The crystal structure shows that Loop 3 is intimately associated with bound biotin (Fig. 2A ▶). The orientation of the side chains of Loop 1 residues also indicate an interaction with bound biotin. The tryptophan residue, W120, which has been shown to contribute substantial energy to the biotin-binding interaction of an adjacent subunit (Sano et al. 1995) is located in Loop 7.
Fig. 1.

Amino acid sequence of streptavidin studied in this paper. Arrows indicate sequences of peptides screened to search for linear epitopes. Loop regions are in capital letters. Sequence starts at residue 13 for core streptavidin.
Fig. 2.
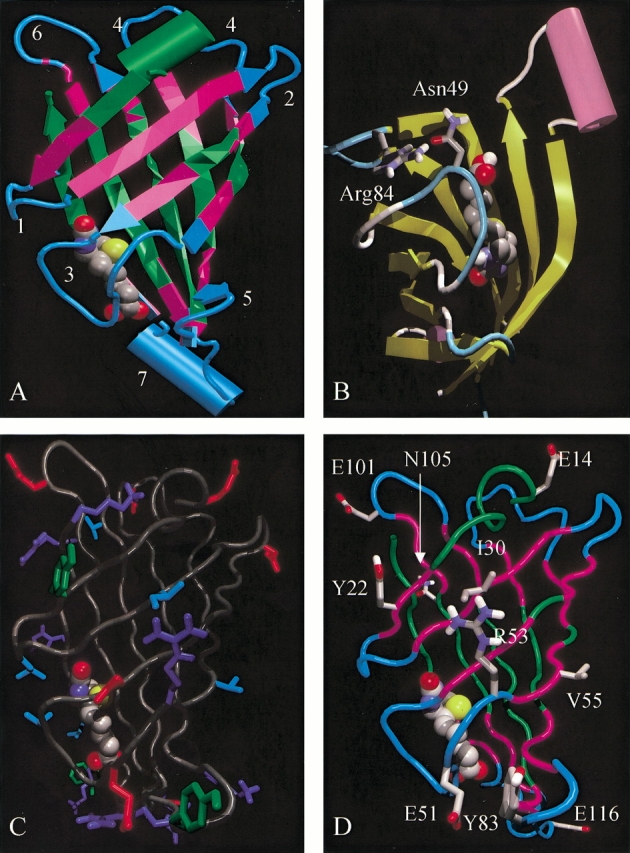
Views of the X-ray crystal structure of streptavidin. (A) Color-coded backbone structure of streptavidin monomer. Cyan, loop regions; magenta, 3-amino acid sequences surrounding loops; green, other regions. Bound biotin is shown in a space-filling rendering, colored by atom type (carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow). The surface loops (1–7) are numbered by the same scheme used for the loop peptides in Fig. 1 ▶. (B) Orientation of side chains of residues R84 (Arg84) and N49 (Asn49). (C) Residues of streptavidin with >30% relative solvent accessible surface area highlighting antibody "interactive" residues. Red, negatively charged residues; dark blue, positively charged residues (including histidine); cyan, surface large hydrophobic residues; green, surface aromatic residues. (D) Streptavidin monomer highlighting residues mutated in Mu37 as defined in Table 2, screen 5. The images were generated using VMD (Humphrey et al. 1996), Raster3D, and Povray (Merritt and Bacon 1997).
Identification of linear epitopes
On the basis of the hypothesis that loop regions make linear epitopes, peptides 1–8 (Fig. 1 ▶) were synthesized and tested in a direct binding assay for recognition by pooled sera from human patients previously immunized with a single dose of murine NR-LU-10 mAb/streptavidin conjugate. Only Peptide 6 was clearly recognized by human IgG (Fig. 3 ▶), which suggested that residue E101, the only charged residue in the exposed loop segment of Peptide 6, was a candidate for mutation. A phage display library confirmed this result but provided no additional information (data not shown).
Fig. 3.

Recognition of peptides 1–8 by pooled human patient serum. Peptides are identified in Fig. 1 ▶ and are shown in order of appearance for each of three dilutions.
Identification of conformational epitopes
Attempts to block the recognition of streptavidin by patient immune serum with Peptide 6 (or the other peptides) were unsuccessful (data not shown). From these results, we assumed that the dominant epitopes are not contained within the peptides and are discontinuous rather than linear.
Screen 1
The initial strategy adopted to identify residues involved in discontinuous immunodominant epitopes was to mutate charged residues in loop regions as conservatively as possible (Table 1). Complementary mutations were made to nonloop residues with opposite charges to maintain the net charge on streptavidin. Mutant (Mu) 1 showed a marked reduction in immunoreactivity with either the pooled patient serum or the murine monoclonal antibody. Mu4, containing an E101Q mutation in Loop 6, showed a minor reduction in immunoreactivity, possibly due to the linear epitope identified by the loop peptide analysis and phage library analysis. Mu2 and Mu3 failed to provide any useful data.
Table 1.
Progressive mutation of streptavidin: Series A
| No. | Mutation | Yield | %IR mu Mab | %IR hu α-sera | Biotin dissociation t1/2, 37°C (min)* |
| Streptavidin | 100 | 100 | 385 | ||
| Screen 1 | |||||
| 1 | E44Q E51Q R53I R84I | + | 37 | 15 | 18 |
| 2 | E14Q D36N R59I K80M | + | 167 | 110 | ND |
| 3 | D67N R103I E116Q H127V | ++ | 100 | 81 | ND |
| 4 | E101Q E116Q K121M K132M | +++ | 58 | 61 | 262 |
| Screen 2 | |||||
| 5 | A46G E51S R53G | + | 5 | 100 | 53 |
| 6 | V97G A100G E101S A102G | − | ND | ND | ND |
| 7 | Deletion: G99 A100 E101 A102 | − | ND | ND | ND |
| 8 | R59G | ± | 100 | 100 | ND |
| 9 | Y83G | + | 8 | 20 | 570 |
| 10 | E51S E116S | ++ | 5 | 76 | ND |
| 11 | E51S | ++ | 6 | 122 | 175 |
| 12 | E51S Y83G | +++ | 1 | 60 | 525 |
| 13 | E44Q E51Q R53I Y83G | − | 2 | 51 | ND |
| 14 | E44Q | +++ | 33 | 44 | 92 |
Abbreviations: mu, murine; hu, human.
* Values are the result of a single determination. Relative standard error is 10% or less except for Mu20, Mu26, and Mu32, for which it is 15% or less.
Mu1 had a significantly increased biotin dissociation rate as compared to native streptavidin (18 and 385 min, respectively; Table 1). Examination of the crystal structure showed an interaction between the side chains of R84 and N49 (Fig. 2B ▶). Previous studies have not described a contribution of this interaction to biotin binding (Sano and Cantor 1990b; Weber et al. 1992a, b; Katz 1997) but mutation of R84 was avoided in subsequent alterations. In contrast to Mu1, Mu4 had a biotin off-rate closer to native streptavidin (262 min; Table 1).
Screen 2
On the basis of the hypothesis that one of the amino acid changes in Mu1 eliminated a binding locus of an immunodominant epitope, further studies were designed to: (1) identify the residues of Mu1 involved in the epitope, (2) explore the extent of the epitope, and (3) identify additional epitopes (Screen 2, Table 1). At this point the constraint of maintaining the net charge was abandoned, because we considered minor charge changes to be of minor influence on the performance of a mutant streptavidin in vivo.
As part of this screen, some mutants were prepared with loops either deleted (e.g., Mu7) or replaced with a loop from a human protein that was known (based on database searching) to have a backbone conformation matching closely that of streptavidin (not shown). These mutants were designed using molecular modeling to ensure that the desired folded structure was energetically and sterically feasible. Nonetheless, the loop-deleted and loop-substituted mutants uniformly resisted proper folding.
Data from screening Mu1 and Mu5 suggested that E51 was part of the epitope recognized by the murine monoclonal antibody. Analysis of Mu11 confirmed this suggestion, but the single E51S mutation failed to reduce the immunoreactivity with pooled patient sera. The X-ray crystal structure showed that the side chain of E51 lies close to the side chain of Y83, one of only two exposed aromatic residues on the surface of streptavidin. Mu9 confirmed the hypothesis that Y83 is part of the epitope recognized by the murine monoclonal antibody, and showed that Y83 is also important to binding with the pooled patient antisera. The Y83 mutation provided the unexpected result that the biotin dissociation rate was reduced by replacement of this tyrosine with glycine (570 min; Table 1). A reduced off-rate was also observed for Mu12, which contains the Y83G mutation in combination with E51S (525 min; Table 2). Molecular modeling has not yet provided an explanation for the impact of the Y83 mutation on the biotin dissociation rate.
Table 2.
Progressive mutation of streptavidin: Series B
| No. | Mutation | Yield | %IR hu α-sera | %IR rb α-19 | Biotin dissociation t1/2, 37°C (min) |
| Streptavidin | NA | 100 | 48 | 385 | |
| Screen 3 | |||||
| 15 | E51S Y83G R53A | +++ | 45 | 423 | |
| 16 | E51S Y83G Y22S | ++ | 48 | 275 | |
| 17 | E51S Y83G E101S | + | 70 | 410 | |
| 18 | E51S Y83G V97A | − | 45 | ND | |
| 19 | E51S Y83G R53A E116S | ± | 8 | 100 | 536 |
| 20 | E51S Y83G Y22S I30A | +++ | 30 | 86 | |
| 21 | E51S Y83G E101S R103S | − | ND | ND | |
| 22 | E51S Y83G V97A V133A | − | ND | ND | |
| 23 | E51S Y83G Y22S V55A I30A | + | 40 | ND | |
| 24 | E51S Y83G E101S R103S D67S | − | 30 | ND | |
| 25 | E51S Y83G V97A V133A N105S | − | ND | ND | |
| 26 | E51S Y83G R53A V55A | + | 38 | 308 | |
| Screen 4 | |||||
| 27 | E51S Y83G R53A E116S V55A | +++ | 6 | 66 | 260 |
| 28 | E51S Y83G R53A E116S D67S | ± | ND | ND | ND |
| 29 | E51S Y83G R53A E116S Y22S | ++ | 8 | 51 | 209 |
| 30 | E51S Y83G R53A E116S N105S | ++ | 8 | 92 | 398 |
| 31 | E51S Y83G R53A E116S V133A | − | ND | ND | ND |
| 32 | E51S Y83G R53A E116S E101S | ++ | 5 | 58 | 493 |
| Screen 5 | |||||
| 33 | E14S Y22S I30S E51S R53A V55A D67S Y83G V97A E101S R103S N105S E116S V133A | − | ND | ND | ND |
| 34 | E14S Y22S I30S E51S R53A V55A D67S Y83G E101S R103S N105S E116S V133A | − | ND | ND | ND |
| 35 | E14S Y22S I30S E51S R53A V55A D67S Y83G E101S N105S E116S V133A | − | ND | ND | ND |
| 36 | E14S Y22S I30S E51S R53A V55A D67S Y83G E101S N105S E116S | − | ND | ND | ND |
| 37 | E14S Y22S I30S E51S R53A V55A Y83G E101S N105S E116S | ++ | 3 | 18 | 73 |
* Values are the result of a single determination. Relative standard error is 10% or less except for Mu20, Mu26, and Mu32, for which it is 15% or less.
Abbreviations: hu, human; rb, rabbit; α-19, anti-Mu19 serum.
Screen 2 results also suggested that residue E44 has an impact on immunoreactivity with either the murine monoclonal antibody or patient antisera. However, mutation of E44 substantially increased the biotin dissociation rate and appeared to have a deleterious effect on folding when mutated in combination with other residues (Mu13), although it was tolerated as a single mutation (Mu14). Therefore, we reasoned that mutation of E51 and Y83 eliminated the main binding interactions for the epitope recognized by the murine monoclonal and that binding at other epitopes, of which E44 might be part, could be reduced by mutating other residues.
Screens 3–5
Having eliminated a major antibody binding locus in Mu12, we returned to the crystal structure of streptavidin to identify as many potential surface interactive residues that could be mutated and still allow proper folding and biotin binding (Fig. 2C ▶). The strategy for Screens 3–5 was modified in several ways. First, a matrix of mutations was mapped out that would elucidate the impact of individual changes on both immune recognition and folding (yield) in a minimum amount of time (Table 2, Screens 3–5). Second, a gene for streptavidin containing all the envisioned mutations (Mu33) was synthesized de novo, so that we could simultaneously work "backward", eliminating "nonpermissive" mutations, identified in Screens 3 and 4, that precluded proper folding of the molecule (Screen 5).
The data in Screens 2, 3, and 4 suggested that E116 (Mu10 vs. Mu11), R53 (Mu12 vs. Mu15), I30 (Mu16 vs. Mu20), Y22 (Mu16 vs. Mu12), V55 (Mu26 vs. Mu15), and E101 (Mu32 vs. Mu19) contributed to some degree to recognition by the pooled patient serum. Mutation of V97, R103, D67, and V133 were found to be nonpermissive, although this characterization is strictly valid only in the context of the other mutations present and for the amino acids to which these residues were changed. Elimination of the four mutations identified as nonpermissive in Screens 3 and 4 from Mu33 (Screen 5) resulted in a readily produced streptavidin mutant. The culmination of this process, Mu37 (Fig. 2D ▶) contained 10 mutations, refolded adequately, and showed a biotin dissociation half-life reduced by ∼80% as compared to native streptavidin (73 vs. 385 min, respectively; Table 2).
In evaluating Mu19, we found immunoreactivity with patient serum was reduced to the point that the effect of further reductions would be difficult to detect. Therefore, rabbits were immunized with Mu19 to generate antiserum similar to anti-streptavidin serum, but lacking reactivity with the dominant epitope. This antiserum was used to address immunoreactivity of mutants in Screens 3, 4, and 5. The murine monoclonal antibody, patient sera pool, and rabbit anti-Mu19 serum only minimally recognized Mu37.
Biochemical analysis of mutants
In addition to yield, recognition by antisera and biotin dissociation rate, mutants were compared to streptavidin in their behavior on SDS-PAGE gels, size exclusion chromatography, and heat stability. Unless boiled, core streptavidin migrates in an SDS-PAGE gel as a 52.4-kD tetramer (Fig. 4 ▶), providing a simple method of determining whether a particular mutant is properly folded. SDS-PAGE analysis of the mutants in Tables 1 and 2 indicated a direct relationship between the number of changes in a mutant and its ability to retain tetrameric structure in the presence of SDS and heat. Streptavidin only partially dissociated from tetramer to monomer when heated in the presence of SDS at 72°C for 1 min, whereas Mu12 completely dissociated under the same conditions. Mu19 and Mu29 completely dissociated at 50°C, and Mu37 completely dissociated at 22°C (Fig. 4 ▶). However, differences in susceptibility to SDS were not reflected in solution stability. Size exclusion HPLC (Fig. 5 ▶) indicated that each of the mutants remained pure tetramer after a 12-h incubation at 37°C in PBS.
Fig. 4.
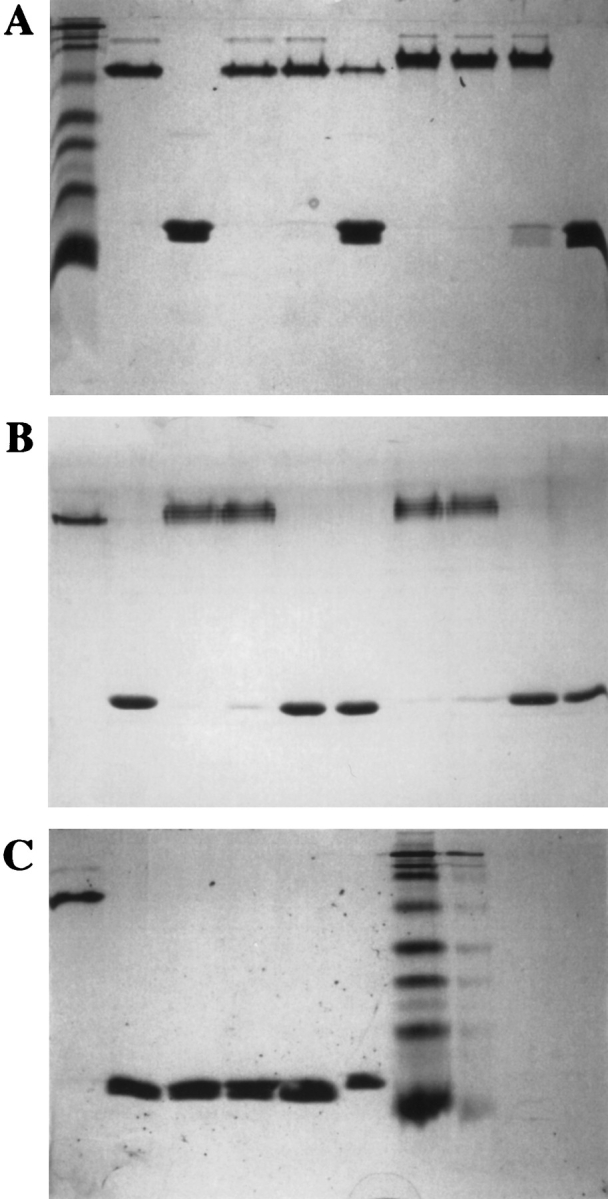
SDS-PAGE analysis of native streptavidin or mutants incubated at various temperatures. (Gel A,) Lane 1, BioRad molecular mass markers: 7.3K, 18.9K, 26.2K, 31.4K, 49.1K, 81.0K, 117K, 211K; lanes 2,3, native streptavidin tetramer (nonboiled) or monomer (boiled in SDS), respectively; lanes 4–6, native streptavidin after treatment at 37°C, 50°C, 72°C, respectively; lanes 7–10, Mu12 after treatment at 22°C, 37°C, 50°C, 72°C, respectively. (Gel B) Lane 1,2, native streptavidin tetramer (nonboiled) or monomer (boiled in SDS), respectively; lanes 3–6, Mu19 after treatment at 22°C, 37°C, 50°C, 72°C, respectively; lanes 7–10, Mu29 after treatment at 22°C, 37°C, 50°C, 72°C, respectively. (Gel C) Lane 1, native streptavidin tetramer (nonboiled); lanes 2–5, Mu37 after treatment at 22°C, 37°C, 50°C, 72°C, respectively; lane 6, native streptavidin monomer (boiled in SDS); lane 7, BioRad molecular mass markers.
Fig. 5.

Size-exclusion high pressure liquid chromatogram of streptavidin (A) and Mu37 (B) after treatment at 37°C. Instability would be indicated by elution of monomer at ∼12 min. Similarly, Mu19 and Mu29 showed no indication of instability in solution at 37°C.
Immunogenicity of selected mutants
Studies were initiated in rabbits to determine whether the decreased immunoreactivity of Mu37 with mAb anti-streptavidin and rabbit anti-Mu19 also resulted in decreased immunogenicity of Mu37. The response to immunization with Mu37 was lower by a statistically significant margin than to native streptavidin (Fig. 6 ▶). IgG response to Mu19 and Mu29 after the second immunization appeared reduced relative to streptavidin, but the difference did not reach the level of statistical significance. The IgM response showed that the diminished IgG levels reflected a reduced overall response rather than an impaired class switch from IgM to IgG (Fig. 6 ▶C,D).
Fig. 6.

Rabbit IgG (A,B) and IgM (C,D) response to a single (A,C) injection of native SA or Mu19, Mu29, or Mu37, or after a repeat injection (B,D) with the same agent. Statistically significant differences are marked (P < 0.05).
To test in vitro the hypothesis that therapy with one form of streptavidin does not limit retreatment with a variant form of streptavidin, the rabbit primary antibody responses to both streptavidin and Mu37 were tested for reactivity to the immunizing antigen and the alternate antigen (Fig. 7 ▶). As shown previously, the increase in IgG titer to Mu37 after a single immunization in incomplete Freund's adjuvant (IFA) was significantly less than the response to a similar injection of streptavidin. In addition, reactivity of anti-streptavidin or anti-Mu37 serum with the alternative antigen was also significantly reduced relative to reactivity with the immunizing antigen.
Fig. 7.

Rabbit anti-SA or anti-Mu37 reactivity with homologous antigen (anti-SA/SA and anti-37/37) or heterologous antigen (anti-SA/37 and anti-37/SA) after a single injection of antigen in IFA in rabbits (n = 4 each). The anti-SA response was statistically greater than the anti-37 response by the Student's t test (P < 0.05).
Discussion
Although the literature describes methods for the identification of epitopes in a wide variety of proteins, there is remarkably little precedent for modulation of protein antigenicity by mutation of these epitopes. The largest body of data in which protein antigenicity is modulated by site-specific mutation, the "humanization" of non-human antibodies, describes the reduction of antigenicity by altering epitopes so that they resemble "self" (Jones et al. 1986; Verhoeyen et al. 1988; Queen et al. 1989; Padlan 1991; Vaswani and Hamilton 1998; Graves et al. 1999), rather than by attempting to reduce the protein's relative antigenicity. Also, there is no analogous strategy for bacterial proteins, like streptavidin, that lack human counterparts. One of the techniques that has been described for humanization of antibodies generated in other animals is "veneering", a process in which surface interactive residues of the structural regions of the antibody are replaced with amino acids from a human antibody sequence. In effect this study represents an attempt to "veneer" a foreign protein.
Collen et al. (1997) have shown that staphylokinase mutants in which mutations of charged residues to Ala are less able to induce antibodies than native staphylokinase (SakSTAR). Mutation of a single Lys residue in this 16-kD protein substantially diminished its ability to induce an immune response in human patients. Laroche et al. (2000) reported production of recombinant SakSTAR variants with reduced antigenicity in human patients using a cumulative mutation strategy very similar to that as we have described here. Neutralizing antibody production developed in 47% of patients treated with SakSTAR variants compared with 81% of patients treated with wild-type SakStar, demonstrating that despite successful reduction in antigenicity there is the problem of individuals showing varying degrees of responses to particular epitopes through the inherent flexibility of the immune response.
A strategy has been outlined (Tiarks et al. 1992) for the reduction of immune response to Factor VIII by replacing epitopes associated with activity-inhibiting antibodies. Porcine/human chimeric Factor VIII retained activity in the presence of inhibiting antibodies to the epitope replaced by the porcine sequence (Lubin et al. 1994). This result is attributable to a difference in immunoreactivity of the chimeric version and not necessarily in antigenicity.
Immunologic properties of streptavidin have been studied. Subramanian and Adiga (1997) performed a detailed comparison of the epitopes of avidin and streptavidin with rabbit antisera. Using the pepscan method of epitope mapping, they identified six linear epitopes in streptavidin and avidin and showed that most of the antibodies to these epitopes are cross-reactive between the two molecules. These results differ from ours in that (1) our patient anti-streptavidin antisera does not react with avidin (data not shown), and (2) relatively little reactivity with linear epitopes is detected in our studies using polyclonal human or goat antisera, although the linear epitope we detected in Loop 6 (Fig. 3 ▶) is among those noted by Subramanian and Adiga (1997). These differences may be attributable to different administration routes (intravenous vs. subcutaneous) and species studied (rabbit vs. human). Furthermore, antiserum raised against the properly folded protein may be distinct from that raised against the denatured antigen.
Streptavidin mutants have been prepared both randomly and site specifically. Chilkoti et al. (1995) have prepared a series of mutants designed to covalently link subunits, reduce the biotin-binding affinity, or allow site-specific derivatization of streptavidin. Chu et al. (1998) engineered a streptavidin mutant in which Loop 3 (our terminology) is deleted and the amino and carboxyl termini are joined through a tetrapeptide linker, generating new protein termini. Remarkably, this protein folds properly and retains a fraction of the biotin-binding affinity of streptavidin. Voss and Skerra (1997) subjected Loop 3 (our terminology) to random mutagenesis to obtain streptavidin mutants with enhanced affinity for a peptide used as a chromatography handle. Sano et al. (1997) designed a dimeric form of streptavidin by site-specifically replacing the energetically favorable π–π interaction of His side chains with a repulsive one by making a H127D mutation and simultaneously deleting Loop 7(our terminology) to reduce the hydrophobicity and preserve the solubility of the dimer. Non-natural photoactive amino acids have also been incorporated into streptavidin in place of the surface-exposed aromatic residue Y83 (Hohsaka et al. 1996).
Because of the paucity of precedent for reduction of antigenicity by mutagenesis, a number of hypotheses were considered in designing the mutants for this study. The first hypothesis was that a large majority of the immune response to streptavidin is directed to a single epitope. After a series of theoretical calculations, Novotny et al. concluded that the majority of the binding energy in antibody–antigen complexes is provided by a small number of residues constituting an energetic epitope (Novotny et al. 1989; Novotny 1991). Thus, residues that could offer high affinity interactions with an antibody were replaced with residues that were less likely to form strong interactions.
Because evidence was found for only a minor response to continuous streptavidin epitopes, site-specific mutation was used in an attempt to identify a dominant conformational epitope. It was determined in Screen 2 that E51 and Y83 could be incorporated into a mutant (Mu12) for which binding to the murine mAb was eliminated, whereas biotin binding and protein folding were maintained. Although mutation of E51 did not decrease the immunoreactivity with pooled patient serum, the strong impact that it had on binding to the murine monoclonal indicated that it could be a major component for antibody recognition. We reasoned that mutation of E51 eliminated the possibility that this site could contribute to the formation of a neo-epitope.
A number of rationales were investigated in the design of mutants in Screen 2. In one strategy, loops were replaced with the (G4S)x linker sequence commonly used in single chain Fv fragments and in fusion proteins (Huston et al. 1988), considered to be flexible and only minimally antigenic. Native residues were retained, however, if according to the crystal structure they appeared to be important for biotin binding or protein structure. In another strategy, databases of human proteins with known three-dimensional structure were searched for loops with α-carbon backbones in conformations closely resembling those of the streptavidin loops. Three of the loops, 1, 2, and 6, fit well with a large number of human protein loops, whereas the other four loops resembled none of the human loops in the databases. The human loops resembling streptavidin loops were then ranked according to a series of parameters indicating how closely the side chain packing and other characteristics resembled those of the streptavidin loops. It was hoped that this strategy would allow proper folding of streptavidin and show reduced immunogenicity. A third strategy involved elimination of loops where β-strands were in close enough proximity to connect through a single glycine residue. None of the examples of human loop replacement or loop elimination yielded functional protein. In the mutants investigated it appeared that multiple mutations grouped together spatially or sequentially were more likely to resist proper folding than mutations that were dispersed over the protein.
The high reactivity of the mutants of Screen 2 with human patient antiserum made it clear that although the epitope recognized by the murine monoclonal antibody is a major component of the human patient antiserum, other epitopes also make substantial contributions. Emphasis was shifted to the replacement of all interactive amino acids with solvent-accessible side chains, regardless of their detectable involvement in patient immune serum recognition. All such residues were identified from the crystal structure (Fig. 2C ▶) and systematically mutated and evaluated as outlined in the Results section. Mu19, Mu29, and Mu37 showed progressively decreasing recognition by patient antiserum and rabbit anti-Mu19 serum. In addition to E51 and Y83, residues including I30, V55, E116, Y22 (the only exposed aromatic residue besides Y83), R53, and E101 may be involved in the binding of patient immune serum to streptavidin. Without crystal structures of the mutants it is impossible to unambiguously conclude that these residues represent epitopes, because their mutation may cause a conformational shift elsewhere in the sequence that is the true recognition site. However, we believe that the structural requirements for the retention of high-affinity biotin binding and proper tetramer formation are strict, and that structural changes substantial enough to markedly alter protein conformation at a site distant from the mutation would also prevent proper folding and diminish biotin binding. Consequently, it is likely that the noted amino acids are, in fact, involved in antibody binding.
Stability of the protein, particularly proteolytic stability, was a major concern. We hypothesized that the mutant protein could be even more antigenic than streptavidin if it were more susceptible to proteolysis and therefore, more effectively processed by antigen-presenting cells. Although little variation was detected in the solution stability of the mutants compared to native streptavidin, their susceptibility to denaturation by heat and SDS did increase in direct relation to the number of changes incorporated. The antigenicity tests did not show a relationship between heat/SDS susceptibility and antigenicity.
The immunoreactivity data in Tables 1 and 2 indicate that Mu19 and Mu37 do not share the residues by which streptavidin binds to human anti-streptavidin immune serum. Testing the antigenicity of these proteins would, therefore, address the question of whether replacement of interactive residues with neutral residues renders the protein less antigenic. Mu37 is less able to elicit an IgG response in rabbits than native streptavidin (Fig. 6A ▶). Mu19 and Mu29 also appear less able to elicit an immune response than streptavidin, but the difference is smaller, and appeared with the response to the second injection and not the first (Fig. 6B ▶). Furthermore, the recognition of streptavidin by Mu37 antiserum is very small (Fig. 7 ▶) indicating that streptavidin epitopes remaining in Mu37 do not become dominant components of the response.
Although the differences between the streptavidin response and the Mu37 response do not represent an alteration in the ability to shift from IgM to IgG, they could represent a difference in ability to mature from low-affinity recognition to high-affinity recognition. The assays used do not differentiate between antibody concentration and affinity. Given that the mutations incorporated were designed to remove residues capable of highly enthalpic interactions, it is entirely possible that the concentration of recognizing antibodies is not substantially changed but that the affinity is reduced. Presumably, either affinity reduction or concentration reduction could accomplish the purpose of allowing multiple injections of the foreign protein for the Pretarget delivery system.
Whether these trends would also occur in human patients is not known. Whether the reduction in immunogenicity observed in rabbits is sufficient to allow repetitive treatments with the pretargeting delivery system is also an open question. More likely, Mu37 and streptavidin could be given as sequential doses in the Pretarget delivery system as the response to one would have little impact on the biodistribution of the subsequent dose. This claim is supported by the data presented in Figure 7 ▶, in which the immunoreactivity of sera from rabbits immunized with either streptavidin or Mu37 failed to bind to the alternative antigen. The data also suggest that the order of administration should be first a conjugate comprised of Mu37 followed by the streptavidin conjugate. In any case, the streptavidin model, like the staphylokinase model (Collen et al. 1997; Laroche et al. 2000), shows that replacement of sufficient residues associated with B-cell epitope binding, with poorly interactive residues, renders a protein not only immunologically distinct from its parent protein, but less antigenic.
Materials and methods
Assay of anti-streptavidin response of immune serum
The immune response to the components of the murine NR-LU-10 mAb/streptavidin conjugate was determined in patients receiving a single dose of the conjugate. Titers of human anti-mouse mAb, human anti-streptavidin, or human anticonjugate were determined by sandwich ELISA.
Serum samples were collected from patients before treatment with mAb/streptavidin conjugate and at weeks 1, 2, 4, 6, 8, 12, and 24 after treatment. Microtiter plates (96-well; Falcon Plastics, Oxnard, CA) were coated with either native, recombinant streptavidin (Roche Molecular Biochemicals, Indianapolis, IN), the murine antibody NR-LU-10 (NeoRx Corp.), or mAb-streptavidin conjugate (NeoRx Corp.), each at a concentration of 1 μg/mL in phosphate buffered saline at pH 7.2 (PBS; BioWhittaker, Walkersville, MD). Plates were incubated for 2 h at ambient temperature and then washed six times in PBS/0.05% Tween 20 (Sigma Chemical Co., St. Louis, MO). All additional reagents were added in PCT buffer (PBS/0.5% Tween 20 plus 5% chicken serum; Sigma Chemical).
A pool of normal human sera (Cutter Biologicals, Cat. # 55510) was used as a standard. Thus, data were calculated in units of normal human serum equivalents (NHS units). After a 1-h incubation at room temperature, and wash, bound antibody was detected with horseradish peroxidase (HRP)-conjugated goat anti-human IgG + IgM (Biosource International, Camarillo, CA) followed by addition of substrate, 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS; Sigma Chemical), in citrate-phosphate buffer for 30 min. Absorbance was read at 415 nm (490 nm reference). Responses were determined by comparing the linear portions of the titration curves for patient and normal human serum.
Immunoreactivity of streptavidin peptides with immune serum
For these studies, streptavidin peptides (Multiple Peptide Systems, San Diego, CA) were coated onto 96-well microtiter plates at 2 μg/mL in PBS. Antiserum with a high anti-streptavidin titer, pooled from seven patients, was then applied to the plates at a dilution of 1:100, 1:200, or 1:400. HRP-conjugated goat anti-human IgG (Biosource International), followed by incubation with ABTS substrate, was used to detect binding.
Construction of the streptavidin mutant genes
Site-directed mutagenesis was performed using the QuickChange kit (Stratagene, La Jolla, CA). All oligonucleotides were synthesized by Operon Technologies (Alameda, CA). Plasmid A140–9, which contained the native core streptavidin in pET3a vector (Novagen, Madison, WI), was used as the starting DNA. The reactions were carried out in 50 μL of supplied reaction mixture containing 400 nM of each oligonucleotide primer, and 50 ng of plasmid DNA. Reactions were performed on a PTC-100 thermal cycler (MJ Research, Inc., Waltham, MA) with 1 cycle of 95°C for 30 sec and 17 cycles of the following profile: 95°C for 30 sec; 55°C for 1 sec; 68°C for 12 sec. The reaction mixture (3 μL) was used to transform Escherichia coli XL-1 Blue competent cells (Stratagene). Plasmid DNAs were prepared from cultures of the transformants using a QIAprep Spin Miniprep kit (Qiagen, Valencia, CA). The sequences were confirmed using a Big Dye DNA sequencing kit (PE Applied Biosystems, Foster City, CA).
Shake flask expression of streptavidin mutants
Cultures of plasmid-containing E. coli BL21 (DE3) pLysS (Novagen) were grown overnight at 37°C in Terrific broth (8 × 500 mL; Sigma Chemical) containing 50 μg/mL of carbenicillin (Sigma Chemical). The cultures were diluted ∼100-fold into fresh media, and isopropyl-β-d-thiogalactopyranoside (0.5 mM; Amersham Pharmacia Biotech Inc., Piscataway, NJ) was added when the cultures attained an optical density (A600) of 0.3–0.6. Incubation was continued for an additional 16 h. Cells were harvested by centrifugation at 4000 g for 10 min at 10°C and washed twice in ice-cold PBS before processing.
Refolding and purification of streptavidin mutants
Cell pellets were resuspended in ice-cold TE (30 mM Tris, 1 mM EDTA at pH 8.0) at 25% w/v. The cells were disrupted through two cycles of microfluidization (M-110S, Microfluidics International, Newton, MA). The insoluble inclusion bodies were recovered by centrifuging the suspension at 15,000 g for 90 min at 10°C and discarding the supernatant.
Recovery of mutants from pelleted inclusion bodies was performed according to Sano and Cantor (1990b). Briefly, a microfluidized pellet mass was resuspended in 8 mL of ice-cold 8 M guanidine-HCl at pH 1.5 per liter of initial cell culture. The resuspension was dialyzed at 5°–10°C against 6 M guanidine-HCl at pH 1.5 to remove endogenous biotin. The dialysate was centrifuged at 10,000 g for 20 min and the supernatant added dropwise to vigorously stirred, ice-cold refolding buffer (4 mM KH2PO4, 16 mM Na2HPO4, 115 mM NaCl at pH 7.0) with a volume at least 31 times that of the supernatant volume. The material was removed, incubated at 4°C overnight without stirring, and spun at 7000 g for 5 min at 10°C. The pellet was discarded.
For iminobiotin affinity purification, the supernatant was adjusted to 50 mM glycine, 500 mM NaCl at pH 9.6, using concentrated stock. Conductivity was adjusted to 46–48 mSiemens/cm with deionized water, and the solution was filtered.
Immobilized iminobiotin (in-house preparation or Pierce Chemical Co., Rockford, IL) for affinity purification was packed in a column and equilibrated in 50 mM glycine, 500 mM NaCl at pH 9. The refolded protein was loaded at room temperature. After washing with 10 bed volumes of 50 mM glycine, 500 mM NaCl at pH 9.6, streptavidin was eluted with 0.2 M sodium acetate, 0.1 M NaCl at pH 4.0. The eluate was neutralized with Tris buffer to give a final concentration of ∼100 mM Tris at pH 7, then exhaustively dialyzed in PBS. Unfolded monomeric streptavidin did not bind to the column.
Yields were assessed qualitatively as recovery from the iminobiotin affinity column. High yields were in the range of 250 mg of purified protein from a 4-L batch, whereas nonpermissive mutants were those that had no recoverable protein from the purification. Samples from each of the purification steps were run on 4%–20% Tris-glycine SDS-polyacrylamide gels (Novex, San Diego, CA) to confirm gene expression and recovery of the tetrameric streptavidin.
Immunoreactivity of streptavidin mutants
Plates were coated with native or mutant streptavidin at 1.0 μg/mL in PBS. After a wash with PBS/Tween buffer, the following detection antibodies were used when indicated: mouse monoclonal anti-streptavidin, rabbit anti-Mu19, and human anti-streptavidin. Bound antibody was detected using HRP-conjugated anti-mouse IgG, anti-rabbit IgG (Jackson Immunoresearch, West Grove, PA), or anti-human IgG (BioSource, Camarillo, CA) as appropriate, and ABTS substrate. The absorbance values of streptavidin-coated wells were compared to absorbance values of wells coated with mutants. The ratio of mutant/streptavidin × 100 is reported as units of percent binding relative to streptavidin.
Biotin dissociation half-life
The rate of biotin dissociation was determined at 37°C in a 1-mL solution of 0.25 M sodium phosphate, 0.15 M NaCl, 0.25% bovine serum albumin (BSA) at pH 7.0, containing 10 μM mutant or native streptavidin, 0.06 μM [3H]biotin (New England Nuclear, Boston, MA; 58 mCi/μmole) and 30 mM ascorbate as [3H]biotin stabilizer. After incubating for 10 min, biocytin (Sigma) was added at 4 mM final concentration (t0).
To assess the extent of [3H]biotin dissociation, aliquots were withdrawn periodically and diluted into PBS containing 0.5% BSA. After mixing, the solution was immediately separated into two equal volumes. To one volume designated total activity, water was added and vortexed. To the second volume, designated free [3H]biotin, equal volumes of 0.2 M ZnSO4 and 0.2 M NaOH were added sequentially to precipitate the protein. After mixing, both fractions were microcentrifuged. Supernatant aliquots (0.15 mL) of each of the designated tubes were then added to scintillation fluid, mixed, and counted in a liquid scintillation counter (Packard Instrument Co., Meriden, CT). Percent radiolabeled biotin bound to native or mutant streptavidin was calculated as cpm of free [3H]biotin/cpm of total activity × 100. Half-lives were calculated from the slopes of standard rate plots.
Temperature/SDS stability of streptavidin mutants
Streptavidin and Mu12, Mu19, Mu29, and Mu37 were maintained at room temperature, 37°C, 50°C, or 72°C for 1 min before application to a 10% Tris buffered SDS-polyacrylamide gel. In addition, streptavidin was maintained at 99°C for 1 min as a positive control for the dissociation of tetramer into monomer. Streptavidin and mutants were also analyzed by size exclusion HPLC on a Zorbax GF-250 column with a 20 mM sodium phosphate/0.5 M NaCl mobile phase and a Varian Dynamax detector set at 280 nm.
Antigenicity of streptavidin mutants in rabbits
Groups of four or five rabbits were immunized subcutaneously with 0.5 mg of streptavidin or Mu19, Mu29, or Mu37 in IFA and 3 wk later in some experiments, the animals were boosted with subcutaneous injection. Sera were collected before and 1 wk after each immunization.
For immune response testing, ELISA plates were coated for 2 h with a 1 μg/mL solution of native or mutant streptavidin in PBS and washed in PBS/0.5% Tween-20. Immune sera from rabbits were typically diluted 1:100, then serially diluted fourfold in PCT. Bound antibody was detected with HRP-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories) followed by ABTS buffer.
A rabbit anti-streptavidin serum was prepared to generate a standard curve. Raw titer values for experimental sera were determined by interpolation into the standard curve. This raw value was divided by the baseline value that resulted from interpolation of absorbance values for preimmune sera into the standard curve. The quotient is reported herein as the titer in units of fold over baseline. This method of reporting the results was used because it allows for direct numerical comparison of the immune responses to similar proteins that vary greatly in their ability to be recognized by a standard antiserum.
Molecular modeling
The effects of mutations of streptavidin were modeled using Macromodel (Schroedinger,Inc., Jersey City, NJ) and Imdad (Molecular Applications Group, Palo Alto, CA). Protein structure–function relationship was determined using Iditis software (Oxford Molecular Ltd., Oxford, UK). The relative solvent accessible surface area was calculated using MC software, which was used to calculate the ratio between the surface area that is able to contact a molecule of the solvent and the maximum solvent-accessible surface area for that residue (Pedersen et al. 1994).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.19901.
References
- Axworthy, D.B., Reno, J.M., Hylarides, M.D., Mallett, R.W., Theodore, L.J., Gustavson, L.M., Su, F-M., Hobson, L.J., Beaumier, P.L., and Fritzberg, A.R. 2000. Cure of human carcinoma xenografts by a single dose of pretargeted yttrium-90 with negligible toxicity. Proc. Natl. Acad. Sci. 97 1802–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitz, H.B., Weiden, P.L., Beaumier, P.L., Axworthy, D.B., Seiler, C., Su, F-M., Graves, S., Bryan, K., and Reno, J.M. 2000. Clinical optimization of pretargeted radioimmunotherapy with antibody streptavidin conjugate and 90Y-DOTA-biotin. J. Nucl. Med. 41 131–140. [PubMed] [Google Scholar]
- Chilkoti, A., Schwarz, B.L., Smith, R.D., Long, C.J., and Stayton, P.S. 1995. Engineered chimeric streptavidin tetramers as novel tools for bioseparations and drug delivery. Bio/Tech. 13 1198–1204. [DOI] [PubMed] [Google Scholar]
- Chu, V., Freitag, S., Le Trong, I., Stenkamp, R.E., and Stayton, P.S. 1998. Thermodynamic and structural consequences of flexible loop deletion by circular permutation in the streptavidin-biotin system. Prot. Sci. 7 848–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collen, D., Stockx, L., Lacroix, H., Suy, R., and Vanderschueren, S. 1997. Recombinant staphylokinase variants with altered immunoreactivity. IV: Identification of variants with reduced antibody induction but intact potency. Circulation 95 463–472. [DOI] [PubMed] [Google Scholar]
- Davis, A.M. and Teague, S.J. 1999. Hydrogen bonding, hydrophobic interactions and the failure of the rigid receptor hypothesis. Angew Chem. Int. Ed. 38 736–749. [DOI] [PubMed] [Google Scholar]
- Delgado, C., Francis, G.E., and Fisher, D. 1992. The uses and properties of PEG-linked proteins. Crit. Rev. Ther. Drug Carrier Syst. 9 249–304. [PubMed] [Google Scholar]
- Fagnani, R., Hagan, M.S., and Bartholomew, R. 1990. Reduction of immunogenicity by covalent modification of murine and rabbit immunoglobulins with oxidized dextrans of low molecular weight. Cancer Res. 50 3638–3645. [PubMed] [Google Scholar]
- Frietag, S., Le Trong, I., Klumb, L., Stayton, P.S., and Stenkamp, R.E. 1997. Structural studies of the streptavidin binding loop. Prot. Sci. 6 1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, D.A., Meares, C.F., David, G.S., McTigue, M., McCall, M.J., Frincke, J.M., Stone, M.R., Bartholomew, R.M., and Leung, J.P. 1986. Monoclonal antibodies as reversible equilibrium carriers of radiopharmaceuticals. Nucl. Med. Biol. 13 383–391. [DOI] [PubMed] [Google Scholar]
- Graves, S.S., Goshorn, S.C., Stone, D.M., Axworthy, D.B., Reno, J.M., Bottino, B., Searle, S., Henry, A., Rees, A.A., and Libby, R.T. 1999. Molecular modeling and preclinical evaluation of the humanized NR-LU-13 antibody. Clin. Cancer Res. 5 899–908. [PubMed] [Google Scholar]
- Hendrickson, W.A., Pahler, A., Smith, J.L., Satow, Y., Merritt, E.A., and Phizackerley, R.P. 1989. Crystal structure of core streptavidin determined from multiwavelength anomalous diffraction of synchrotron radiation. Proc. Natl. Acad. Sci. 86 2190–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohsaka, T., Ashizuka, Y., Murakami, H., and Sisido, M. 1996. Incorporation of nonnatural amino acids into streptavidin through in vitro frame-shift suppression. J. Am. Chem. Soc. 118 9778–9779. [Google Scholar]
- Humphrey, W., Dalke, A., and Schulten, K. 1996. VMD—Visual molecular dynamics. J. Molec. Graphics 14.1 33–38. [DOI] [PubMed] [Google Scholar]
- Huston, J.S., Levinson, D., Mudgett-Hunter, M., Tai, M-S., Novotny, J., Margolies, M.N., Ridge, R.J., Bruccoleri, R.E., Haber, E., Crea, R., and Oppermann, H. 1988. Protein engineering of antibody binding sites: Recovery of an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. 85 5879–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P.T., Dear, P.H., Foote, J., Neuberger, M.S., and Winter, G. 1986. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321 522–535. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Katz, B.A. 1997. Binding of biotin to streptavidin stabilizes intersubunit salt bridges between Asp61 and His87 at low pH. J. Mol. Biol. 274 776–800. [DOI] [PubMed] [Google Scholar]
- Knox, S.J., Goris, M.L., Weiner, L.M., Tempero, M., Weiden, P.L., Gentner, L., Breitz, H., Axworthy, D., Seiler, C., Bryan, K., Fisher, D.R., and Horak, I.D. 2000. Phase II trial of yttrium-90-DOTA-biotin pretargeted by NR-LU-10 antibody/streptavidin in patients with metastatic colon cancer. Clin. Cancer Res. 6 406–414. [PubMed] [Google Scholar]
- Kurzban, G.P., Bayer, E.A., Wilchek, M., and Horowitz, P.M. 1991. The quaternary structure of streptavidin in urea. J. Biol. Chem. 266 14470–14474. [PubMed] [Google Scholar]
- Laroche, Y., Heymans, S., Capaert, S., De Cook, F., Demarsin, E., and Collen, D. 2000. Recombinant staphylokinase variants with reduced antigenicity due to elimination of B-lymphocyte epitopes. Blood 96 1425–1432. [PubMed] [Google Scholar]
- Lubin, I.M., Healey, J.F., Scandella, D., Runge, M.S., and Lollar, P. 1994. Elimination of a major inhibitor epitope in factor VIII. J. Biol. Chem. 269 8639–8641. [PubMed] [Google Scholar]
- Marshall, D., Pedley, R.B., Boden, J.A., Melton, R.G., and Begent, R.H. 1996. Polyethylene glycol modification of a galactosylated streptavidin clearing agent: Effects in immunogenicity and clearance of a biotinylated anti-tumor antibody. Br. J. Cancer 73 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton, R.G., Searle, F., Sherwood, R.F., Bagshawe, K.D., and Boden, J.A. 1990. The potential of carboxypeptidase G2: Antibody conjugates as anti-tumor agents. II. In vivo localising and clearance properties in a choriocarcinoma model. Br. J. Cancer 61 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Meyer, D.L., Jungheim, L.N., Law, K.L., Mikolajczyk, S.D., Shepherd, T.A., Mackensen, D.G., Briggs, S.L., and Starling, J.J. 1993. Site-specific prodrug activation by antibody-β-lactamase conjugates: Regression and long term growth inhibition of human colon carcinoma xenograft models. Cancer Res. 53 3956–3963. [PubMed] [Google Scholar]
- Mikolajczyk, S.D., Meyer, D.L., Fagnani, R., Hagan, M.S., Law, K.L., and Starling, J.J. 1996. Dextran modification of Fab`-β-lactamase conjugate modulated by variable pretreatment of Fab` with amine-blocking reagents. Bioconj. Chem. 7 150–158. [DOI] [PubMed] [Google Scholar]
- Novotny, J. 1991. Protein antigenicity: A thermodynamic approach. Mol. Immunol. 28 201–207. [DOI] [PubMed] [Google Scholar]
- Novotny, J., Bruccoleri, R.E., and Saul, F.A. 1989. On the attribution of binding energy in antigen/antibody complexes McPc603, D1.3, and HyHEL-5. Biochem. 28 4735–4749. [DOI] [PubMed] [Google Scholar]
- Padlan, E.A. 1991. A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand-binding properties. Mol. Immunol. 28 489–498. [DOI] [PubMed] [Google Scholar]
- Pahler, A., Hendrickson, W.A., Kolks, M., Argarana, C.E., and Cantor, C.R. 1987. Characterization and crystallization of core streptavidin. J. Biol. Chem. 262 13933–13937. [PubMed] [Google Scholar]
- Pedersen, J.T., Henry, A.H., Searle, S.J., Guild, B.C., Roguska, M., and Rees, A.R. 1994. Comparison of surface accessible residues in human and murine immunoglobulin Fv domains. Implication for humanization of murine antibodies. J. Mol. Biol. 235 959–973. [DOI] [PubMed] [Google Scholar]
- Queen, C., Schneider, W.P., Selick, H.E., Payne, P.W., Levitt, M., Junghans, R.P., and Waldmann, T.A. 1989. A humanized antibody that binds to the Interleukin-2 receptor. Proc. Nat. Acad. Sci. 86 10029–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano, T. and Cantor, C.R. 1990a. Cooperative biotin binding by streptavidin. electrophoretic behavior and subunit association of streptavidin in the presence of 6M urea. J. Biol. Chem. 265 3369–3373. [PubMed] [Google Scholar]
- ———. 1990b. Expression of a cloned streptavidin gene in Escherichia coli. Proc. Natl. Acad. Sci. 87 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1995. Intersubunit contacts made by tryptophan 120 with biotin are essential for both strong biotin binding and biotin-induced tighter subunit association of streptavidin. Proc. Natl. Acad. Sci. 91 3180–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano, T., Pandori, M.W., Chen, X., Smith, C.L., and Cantor, C.R. 1995. Recombinant core streptavidins. A minimum-sized core streptavidin has enhanced structural stability and higher accessibility to biotinylated macromolecules. J. Biol. Chem. 270 28204–28209. [DOI] [PubMed] [Google Scholar]
- Sano, T., Vajda, S., Smith, C.L., and Cantor, C.R. 1997. Engineering subunit association of multisubunit proteins: A dimeric streptavidin. Proc. Natl. Acad. Sci. 94 6153–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, J., Lin, Y., Sanderson, J., Zuo, Y., Stone, D., Mallett, R., Wilbert, S., and Axworthy, D. 2000. A tetravalent single chain antibody-streptavidin fusion protein for pretargeted lymphoma therapy. Cancer Res. 60 6663– 6669. [PubMed] [Google Scholar]
- Senter, P.D. 1990. Activation of prodrugs by antibody–enzyme conjugates: A new approach to cancer therapy. FASEB J. 4 188–193. [DOI] [PubMed] [Google Scholar]
- Subramanian, N. and Adiga, P.R. 1997. Mapping the common antigenic determinants in avidin and streptavidin. Biochem. Mol. Biol. Int. 43 375–382. [DOI] [PubMed] [Google Scholar]
- Tamura, M., Milenic, D.E., Iwahashi, M., Padlan, E., Schlom, J., and Kashmiri, S.V.S. 2000. Structural correlates of an anticarcinoma antibody: Identification of specificity-determining residues (SDRs) and development of a minimally immunogenic antibody variant by retention of SDRs only. J. Immunol. 164 1432–1441. [DOI] [PubMed] [Google Scholar]
- Tiarks, C., Humphreys, R.E., Anderson, J., Mole, J., and Pechet, L. 1992. Hypothesis for the control of clotting Factor VIII inhibitory antibodies by decreasing potency of helper T-cell-recognized epitopes in Factor VIII. Scand. J. Immunol. 36 653–660. [DOI] [PubMed] [Google Scholar]
- Vaswani, S.K. and Hamilton, R.G. 1998. Humanized antibodies as potential therapeutic drugs. Ann. Allergy Asthma Immunol. 81 105–116. [DOI] [PubMed] [Google Scholar]
- Verhoeyen, M., Milstein, C., and Winter, G. 1988. Reshaping human antibodies: Grafting an antilysozyme activity. Science 239 1534–1536. [DOI] [PubMed] [Google Scholar]
- Voss, S. and Skerra, A. 1997. Mutagenesis of a flexible loop in streptavidin leads to higher affinity for the Strep-tag II peptide and improved performance in recombinant protein purification. Prot. Eng. 10 975–982. [DOI] [PubMed] [Google Scholar]
- Wawrzynczak, E.J. 1991. Systemic immunotoxin therapy of cancer: Advances and prospects. Br. J. Cancer 64 624–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, P.C., Ohlendorf, D.H., Wendodloski, J.J., and Salemme, F.R. 1992a. Structural origins of high-affinity biotin binding to streptavidin. Science 234 85–88. [DOI] [PubMed] [Google Scholar]
- Weber, P.C., Wendodloski, J.J., Pantoliano, M.W., and Salemme, F.R. 1992b. Crystallographic and thermodynamic comparison of natural and synthetic ligands bound to streptavidin. J. Am. Chem. Soc. 114 3197–3200. [Google Scholar]
- Weber, P.C., Pantoliano, M.W., Simons, D.M., and Salemme, F.R. 1994. Structure-based design of synthetic azobenzene ligands for streptavidin. J. Am. Chem. Soc. 116 2717–2724. [Google Scholar]
- Zamdidis, E.T. and Scott, D.W. 1996. Epitope-specific tolerance induction with an engineered immunoglobulin. Proc. Natl. Acad. Sci. 93 5019–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]