

Abstract
A collection of circularly permuted catalytic chains of aspartate transcarbamoylase (ATCase) has been generated by random circular permutation of the pyrB gene. From the library of ATCases containing permuted polypeptide chains, we have chosen for further investigation nine ATCase variants whose catalytic chains have termini located within or close to an α helix. All of the variants fold and assemble into dodecameric holoenzymes with similar sedimentation coefficients and slightly reduced thermal stabilities. Those variants disrupted within three different helical regions in the wild-type structure show no detectable enzyme activity and no apparent binding of the bisubstrate analog N-phosphonacetyl-l-aspartate. In contrast, two variants whose termini are just within or adjacent to other α helices are catalytically active and allosteric. As expected, helical disruptions are more destabilizing than loop disruptions. Nonetheless, some catalytic chains lacking continuity within helical regions can assemble into stable holoenzymes comprising six catalytic and six regulatory chains. For seven of the variants, continuity within the helices in the catalytic chains is important for enzyme activity but not necessary for proper folding, assembly, and stability of the holoenzyme.
Keywords: Circular permutation, protein engineering, cooperativity, folding, stability
Circular permutation of polypeptide chains has been exploited to examine the relationship between the amino acid sequence of a protein and its properties, including structure, stability, and function. In contrast to site-specific substitution of amino acids, circular permutation changes the connectivity within the polypeptide chain without altering the amino acid composition. At the level of primary structure, permutation produces a rearranged polypeptide that starts at the newly introduced N terminus somewhere within the wild-type sequence, continues through the chain from the C to the N terminus through an appropriate linker, and ends at the residue just before the new starting position. As a result, the normal (wild-type) termini of a polypeptide are linked, and new termini are introduced elsewhere in the three-dimensional structure.
Since many proteins have their N and C termini located in close proximity in the three-dimensional structure, it is possible to link the terminal regions and introduce new termini elsewhere, thereby forming circularly permuted chains. More than a dozen proteins have been permuted to date (for review, see Heinemann and Hahn 1995). These include bovine pancreatic trypsin inhibitor (Goldenberg and Creighton 1983), phosphoribosylanthranilate isomerase (Luger et al. 1989), T4 lysozyme (Zhang et al. 1993), dihydrofolate reductase (Buchwalder et al. 1992; Uversky et al. 1996; Nakamura and Iwakura 1999), chymotrypsin inhibitor 2 (Otzen and Fersht 1998), barnase (Tsuji et al. 1999), ribonuclease T1 (Mullins et al. 1994; Garrett et al. 1996; Johnson and Raushel 1996), streptavidin (Chu et al. 1998), β-lactamase (Pieper et al. 1997), βB2-crystallin (Wieligmann et al. 1998), Dsb A (Hennecke et al. 1999), and the catalytic (c) polypeptide chain of Escherichia coli aspartate transcarbamoylase (aspartate carbamoyltransferase, carbamoyl phosphate: l-aspartate carbamoyltransferase, EC 2.1.3.2, ATCase) (Yang and Schachman 1993; Graf and Schachman 1996; Zhang and Schachman 1996). Surprisingly, the major rearrangements in the order of the secondary structural elements resulting from the altered primary sequence often produce proteins that are similar to their wild-type counterparts both catalytically and structurally, as demonstrated by two-dimensional NMR spectroscopy (Zhang et al. 1993; Viguera et al. 1995) and X-ray crystallography (Hahn et al. 1994; Viguera et al. 1996; Pieper et al. 1997; Chu et al. 1998).
Several general conclusions have been drawn from these studies. First, circular permutation often results in proper folding in vivo; it is therefore not necessary that the polypeptide chain be synthesized in a specific temporal order for proper folding. Second, termini may be positioned in many or all loops in a protein, as shown for the SH3 domain (Viguera et al. 1996) and RNaseT1 (Garrett et al. 1996). Third, continuity of the chain within a domain is not essential for proper nucleation of protein folding, as demonstrated by permutation of the T4 lysozyme (Zhang et al. 1993) and the c chain of ATCase (Yang and Schachman 1993; Zhang and Schachman 1996), both of which have two structural domains. A final conclusion is that proteins are surprisingly tolerant to circular permutation, because the new termini of several proteins have been placed in a variety of locations. However, an exhaustive study of the tolerance for locating chain termini throughout the protein structure has yet to be reported.
Toward this goal, a method has been developed for random circular permutation of genes and expressed polypeptide chains (Graf and Schachman 1996). In brief, this method involves (1) circularization of an exact open reading frame; (2) random, limited cleavage with DNase I; (3) repair of the frayed ends of the polynucleotide chains; (4) incorporation of the resulting linear, double-stranded fragments into a suitable expression plasmid; and (5) screening for stable, active protein. In the case of ATCase, the variants with permuted c chains were identified by selecting for activity by suppression of auxotrophy and for protein production by an immunoblot screen (Graf and Schachman 1996).
Many questions regarding the rules for positioning chain termini can be addressed using this approach. Can the ends be placed within elements of secondary structure or in the hydrophobic core? Are there regions of the structure where continuity of the polypeptide chain is essential for folding, assembly, or stability of proteins? Are functional properties including catalysis and regulation of enzyme activity dependent on the location of ends? How widely dispersed throughout the structure can the termini be located? Random circular permutation, as applied to the c chain of ATCase, provides a direct approach to address these questions.
For many purposes, the structural and functional complexity of ATCase is advantageous because the effects of the placement of termini on different properties of the enzyme can be assessed. Catalytic chains assemble into stable trimers (c3) that combine with regulatory (r) dimers (r2) to form ATCase holoenzyme (c6r6). Thus, the effect of the terminal position of c chains can be assessed with regard to the stability of the enzyme or the catalytic trimer. Moreover, the function of the enzyme can be assessed in terms of enzyme activity and homotropic and heterotropic allosteric effects. Therefore, it may be possible to identify regions of the polypeptide important for different aspects of protein function.
Rationale for choice of mutants
For most circularly permuted proteins generated by site-directed techniques, the newly introduced termini were located in flexible, solvent-exposed loops or turns in the polypeptide chains. Although the c chain of ATCase has been permuted at many positions, only two constructs have been produced to examine the effect on enzyme activity of placing termini in buried regions (Zhang and Schachman 1996). Both of these permuted c chains possessed termini in β strands, one variant in each of the two structural domains. One of these constructs produced enzyme of low activity in good yield, whereas the second resulted in no detectable protein or enzyme activity. The random permutation method was developed to address more exhaustively whether chain termini can be located in elements of secondary structure and buried regions, without the inherent bias of choosing a permutation site. More than 30 permutants have been generated to date (Graf and Schachman 1996; Y.R. Yang and H.K. Schachman, unpubl.). A topological map of the c chain in Figure 1 ▶ shows the α helices and β strands along with the location of newly introduced N termini. It is worth noting that, in contrast to the earlier map (Graf and Schachman 1996), the termini in these permutants are distributed through the structure with almost equal numbers located in the carbamoyl phosphate and aspartate domains. Those permutants designated by dashed arrows were identified only by DNA sequence determinations, whereas those indicated by solid arrows have been purified and are the subject of the investigations reported here.
Fig. 1.

Circular topological map of the c chain in ATCase along with location and identification of N and C termini in the circularly permuted chains. Secondary structural elements—β strands and α helices—are designated along the circle as red and purple boxes, respectively. The carbamoyl phosphate-binding domain contains residues 1–134, and the aspartate-binding domain includes residues 150–284. Random permutants were identified by the immunoblot assay or by suppression of auxotrophy owing to the presence of active enzyme in the colonies. Those permutants for which the complete DNA sequences were obtained are designated by dashed arrows, with the N-terminal residue indicated by the one-letter code and the number of the residue based on the wild-type sequence. The slash (/) indicates the bulk of the protein sequence, followed by the determined C-terminal region, either a single amino acid residue with the numbered position or additional residues resulting from the use of stop codons in the second or third reading frame. Permutants designated by solid arrows represent those proteins that have been purified and characterized. The topological map is based on the crystallographic structure (Ke et al. 1988) and coordinates deposited in the Protein Data Bank (registry number 6AT1). Labeling of α helices is taken from Lipscomb (1994). Nomenclature is according to Graf and Schachman (1996). The N-terminal residue (excluding initiator Met) and its position in the wild-type ATCase sequence are given, followed by the last residue of the c chain and its position, then any extra residues encoded by the triple frame stop codons. Accordingly, the wild-type sequence would be denoted as A1/L310.
The proteins selected for the present study were those having termini located within or near helical regions. Some of the permuted proteins are inactive because the introduction of the N and C termini and the loss of continuity of the polypeptide chain in that vicinity lead to destruction of the active site. Nonetheless these polypeptide chains apparently fold into the secondary and tertiary structures required for the formation of trimers and association with regulatory dimers to yield stable holoenzymes. Figure 2 ▶ shows the locations of the newly introduced N termini in relation to the helical regions and the active-site cleft between the two domains. Two variants had N termini at adjacent positions, residues V169 and H170, in Helix 6. Others started at the same position, residue Q137, within Helix 5, but were 5 residues shorter or 4 residues longer than the wild-type c chain (Graf and Schachman 1996). For comparison, an exactly permuted sequence starting at the same position was generated to examine the effects of small insertions and deletions that often result from the random permutation method. Additionally, a construct with termini immediately adjacent to a helix, as in A32, was compared with an enzyme comprising c chains with an interrupted α helix, as in R56. The permuted variants were investigated using biochemical and biophysical methods to assess the effects of disrupting α helices of the c chains on the stability, catalytic, and regulatory properties of the holoenzyme.
Fig. 2.

Ribbon representation of the wild-type c chain of ATCase. The position of N termini in permuted variants is represented by the junctions between colors, with numbering based on the wild-type sequence. In the permuted variants, the N and C termini of the wild-type enzyme are connected through a six- or eight-residue linker. The N-terminal domain comprising residues 1–134 (in purple) is on the left, and the C-terminal domain composed of residues 150–284 (in green and red) is on the right. The two domains are linked covalently by Helix 5 (in blue), comprising residues 135–149. Helix 12 (in green), comprising residues 285–305, crosses back from the C-terminal domain and interacts substantially with residues in the N-terminal domain. The locations of the new termini in specific helices are designated in the topological map (Fig. 1 ▶). The figure was generated using MolScript (Kraulis 1991) with one c chain from the crystal structure of the unliganded holoenzyme (Ke et al. 1988).
Results
Primary structure of permuted c chains
Eight circularly permuted variants, with c chains shown schematically in Figures 1 and 2 ▶ ▶, have termini positioned in α-helical regions of the wild-type enzyme, leading to the interruption of Helix 2, Helix 5, Helix 6, and Helix 8. In the two randomly produced variants lacking continuity in Helix 6, the N termini are at adjacent residues, and there are additional residues incorporated at the C termini leading to polypeptide chains longer than the wild-type chains. In contrast, the variants with interruptions in Helix 5 have the same N terminus but end at different positions, with one variant having a significant deletion and the other an insertion. One other variant, A32/A32-HKVN, has an N terminus immediately adjacent to the C terminus of Helix 1, whereas I215/E217-VR has the N terminus just inside the N-terminal region of Helix 8. These variants are useful for comparing proteins with permutation sites within helices to those having disruptions outside an α helix.
Electrospray ionization mass spectrometry was performed on all the purified variants in order to determine whether the DNA sequence was a reliable indicator of the resulting structure. The resulting molecular mass determinations confirmed that no secondary mutations were introduced by the random permutation technique, and they also indicated the degree of processing of the N-terminal Met. As seen in Table 1, the observed masses of the permuted c chains are in excellent agreement with the values based on the DNA sequence. Although the wild-type c chain is totally devoid of the N-terminal Met, only one of the permuted variants, A32/A32-NKVN, undergoes full N-terminal processing. The H169/S171-LR variant displays partial processing, and the remaining seven variants are not processed.
Table 1.
Mass spectromic analysis of ATCase c chain variants
| Variant | MVcalc (with Met) | MWcalc (without Met) | MWobs | N-terminal processing (%) |
| A32/A32-NKVNa | 35587.81 | 35456.62 | 35457 | >95 |
| Q137/S131 | 34483.63 | 34352.44 | 34484 | <5 |
| Q137/T136 | 35061.16 | 34929.27 | 35063 | <5 |
| Q137/T136-HKVN | 35539.71 | 35408.52 | 35544 | <5 |
| V169/S171-LR | 35653.93 | 35522.74 | 35522 | ∼50 |
| 35653 | ||||
| H170/T173-HKVN | 35978.26 | 35847.07 | 35980 | <5 |
| I215/E217-VR | 35687.87 | 35556.68 | 35688 | <5 |
| R54/T53-LRb | 35664.67 | 35533.48 | 35664 | <5 |
| R56/R54-IRb | 35563.56 | 35432.37 | 35564 | <5 |
a Nomenclature is according to Graf and Schachman (1996). The N-terminal residue (excluding initiator Met) and its position in the wild-type c chain sequence are given, followed by the last residue and its position, then any extra residues encoded by the triple frame stop codons. Thus, the wild-type sequence would be denoted as A1/L310.
b Linker to express these chains included GSHHHHHH, which differs from that, SGELDM, reported earlier (Yang and Schachman 1993; Graf and Schachman 1996).
Quaternary structure and activity of holoenzymes containing permuted c chains
Electrophoresis of seven purified variants in nondenaturing polyacrylamide gels stained for protein (Fig. 3 ▶) demonstrated a high degree of purity (>95%) for each of the holoenzymes with mobilities very similar to that of wild-type ATCase (Fig. 3 ▶, lane 1). Reversed-phase HPLC and mass spectrometry provided independent evidence confirming the purity of the subunits isolated from the variants. Analogous electrophoresis experiments on polyacrylamide gels stained for enzyme activity showed single bands for the variants I215/E217-VR and A32/A32-NKVN, but no activity for the remaining permuted variants (data not shown). The sedimentation coefficients of holoenzymes containing permuted c chains were similar to that of wild-type ATCase (11.5 S), ranging from 11.3 S to 11.6 S. Some of the holoenzymes, such as those with N termini in the c chains at R54 and R56 as well as those starting at Q137, proved substantially less stable than the others, as shown by both sedimentation velocity and electrophoresis experiments. For these variants dissociation into catalytic trimers and regulatory dimers was readily apparent.
Fig. 3.

Characterization of holoenzymes containing circularly permuted c chains by polyacrylamide gel electrophoresis under nondenaturing conditions. Proteins (10 μg/lane) were separated on a 7.5% polyacrylamide gel and stained with Coomassie brilliant blue G-250. (Lane 1) ATCasewt; (Lane 2) c trimer; (Lane 3) ATCaseH6; (Lane 4) A32/A32-NKVN; (Lane 5) Q137/S131; (Lane 6) Q137/T136; (Lane 7) Q137/T136-HKVN; (Lane 8) H169/S171-LR; (Lane 9) H170/T173-HKVN; (Lane 10) I215/E217-VR.
Enzyme kinetics of variants containing circularly permuted c chains
Of the nine ATCases containing circularly permuted c chains, only two exhibited significant enzyme activity. The variant A32/A32-NKVN, whose termini are close to the C terminus of Helix 1, had a Vmax about 31% and a K0.5 about 27% those of ATCasewt (Table 2). Since all of the variants with circularly permuted c chains also contained regulatory chains with a six-residue His-tag at the N terminus, the parameters for ATCaseH6 are shown for comparison. As seen in Table 2, the presence of the His-tag had virtually no effect on the enzyme activity of the holoenzyme. The kinetic parameters for the I215/E217-VR variant were very similar to those of the wild-type enzyme. Both variants exhibited a sigmoidal dependence of enzyme activity on aspartate concentration, as shown by the marked curvature in the Eadie–Hofstee plots (Fig. 4 ▶B,C). The homotropic cooperativity with respect to aspartate, as revealed by the Hill coefficient, was 1.6 for the A32/A32-NKVN variant and 1.3 for the I215/E217-VR variant, as compared to 1.7 for ATCasewt and 1.6 for ATCaseH6.
Table 2.
Kinetic parameters of ATCase holoenzyme variants containing circularly permuted c chainsa
| Variant | K0.5 (mM Asp) | Vmax (mmol • mg−1 • h−1) | nH |
| ATCasewt | 7.5 | 10.8 | 1.7 |
| ATCaseH6 | 6.2 | 9.8 | 1.6 |
| A32/A32-NKVN | 2.0 | 3.4 | 1.6 |
| I215/E217-VR | 6.5 | 9.0 | 1.3 |
a The mutant proteins not listed here showed no detectable activity (<0.1%) in enzyme assays with 30–600 μg purified protein or following a sensitive activity stain of nondenaturing polyacrylamide gels loaded with 10 μg of the mutant proteins. The randomly generated clones (not listed) also failed to suppress pyrimidine auxotrophy in a highly sensitive complementation assay.
Fig. 4.
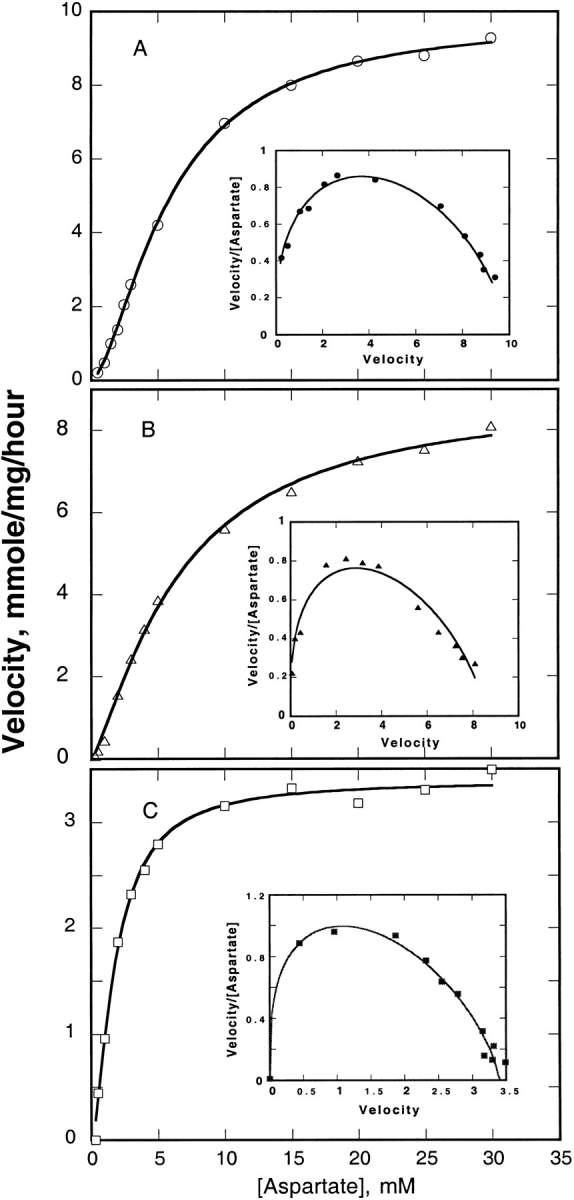
Enzyme activity of ATCase variants. Enzyme assays were performed at varying aspartate concentrations as described in Materials and Methods. The solid line represents a fit of the data to the Hill equation to obtain the kinetic parameters given in Table 2. Inset figures show Eadie–Hoftsee plots to illustrate the degree of cooperativity. (A) ATCaseH6, (B) I215/E217-VR, (C) A32/A32-NKVN.
As seen from the sigmoidal saturation plots, the two active variants show the homotropic allosteric effects characteristic of ATCasewt. The sharp plateau of velocity for the A32/A32-NKVN variant (Fig. 3C ▶) indicates substrate inhibition above 10-mM aspartate. The active permuted enzymes exhibited allosteric activation and inhibition, respectively, by the effectors ATP and CTP (data not shown), indicating that they retain the heterotropic regulation characteristic of wild-type ATCase. The remaining variants were estimated from enzyme assays to possess <0.1% of the activity of wild-type enzyme and also failed to suppress pyrimidine auxotrophy in a highly sensitive complementation assay.
PALA binding and associated conformational changes
Since seven of the ATCase variants exhibited no detectable catalytic activity, it was important to determine whether these permutants were capable of binding the bisubstrate analog PALA, which is bound to the wild-type enzyme with much higher affinity (K0.5 = 105 nM) than are the substrates carbamoyl phosphate (K0.5 = 0.2 mM) and aspartate (K0.5 = 8 mM). Accordingly, the binding of PALA to the variants was assessed by difference spectroscopy (Collins and Stark 1971). As seen in Table 3, the values for the spectral change caused by PALA, ΔA290–285 of 0.014 and 0.011 cm−1 • mg−1 • mL for the ATCasewt and ATCaseH6, respectively, were very similar to that (0.013 cm−1 • mg−1 • mL) measured previously (Howlett and Schachman 1977). Similarly, the active variants A32/A32-NKVN and I215/E217-VR exhibited analogous difference spectra with magnitudes of 0.011 and 0.008 cm−1 • mg−1 • mL, respectively (Table 3). In contrast, none of the inactive mutants showed a significant change in the absorption spectrum (Table 3), with values of ΔA290–285 less than 0.001 cm−1 • mg−1 • mL. Even at a 10-fold higher concentration of PALA (40 PALA/active site), there was no effect on the ultraviolet absorption spectra of these inactive variants.
Table 3.
Physical properties of ATCase holoenzyme variants containing circularly permuted c chains
| Variant | s (S) | Δs/sa,b (%) | Tma,c (°C) | ΔTmd (°C) | ΔA290-285e (mg • cm • mL−1 |
| ATCasewt | 11.5 | −2.9 | 64.1 | +0.6 | 0.014 |
| ATCaseH6 | 11.6 | −2.6 | 63.5 | +0.0 | 0.011 |
| A32/A32-NKVN | 11.6 | −2.0 | 62.5 | −1.0 | 0.011 |
| Q137/S131 | 11.4 | −0.0 | 59.8 | −3.7 | 0.000 |
| Q137/T136 | 11.5 | n.d.f | 49.7 | −13.8 | 0.001 |
| Q137/T136-HKVN | 11.6 | n.d.f | 49.6 | −13.9 | 0.001 |
| V169/S171-LR | 11.4 | +0.2 | 62.8 | −0.7 | 0.001 |
| H170/T173-HKVN | 11.5 | −0.1 | 61.7 | −1.8 | 0.001 |
| I215/E217-VR | 11.4 | −2.3 | 59.9 | −3.6 | 0.008 |
| R54/T53-LR | 11.4 | +0.3 | 64.5 | +1.0 | n.d.f |
| R56/R54-IR | 11.3 | −0.2 | 59.4 | −4.1 | n.d.f |
a Average of two or more experiments performed on two independent protein preparations.
b Percent change in the sedimentation coefficient promoted by the bisubstrate analog PALA.
cTm is the midpoint of the unfolding transition in differential scanning calorimetry experiments.
d Difference in Tm relative to ATCaseH6.
e ΔA290-285 is the absorbance difference between the maximum at 290 nm and the minimum at 285 nm for protein containing PALA (4 PALA/active site) versus without PALA.
f Not determined.
In addition to binding with high affinity to wild-type ATCase, PALA is known to promote the global conformational change in the enzyme associated with its allosteric properties (Howlett and Schachman 1977). Studies with many mutant forms of ATCase have demonstrated that those holoenzymes that exhibit cooperativity in enzyme kinetics or the binding of analogs also undergo a structural change revealed by a 3% decrease in sedimentation coefficient (s). Results of the difference sedimentation velocity experiments (Δs/s, in percent) in the absence and presence of PALA are summarized in Table 3. The values for the wild-type enzyme and for ATCaseH6, −2.9% and −2.6%, respectively, are in good agreement with that (−3.1%) obtained previously (Howlett and Schachman 1977). Catalytically active holoenzymes containing permuted c chains yielded Δs/s values that were slightly smaller in magnitude (−2.0% for A32/A32-NKVN and −2.3% for I215/E217-VR). In contrast, the inactive variants Q137/T136, V169/S171-LR, H170/T173-HKVN, R54/T53-LR, and R56/R54-IR showed no detectable change in the sedimentation coefficient upon the addition of PALA (Table 3).
Thermal stability of ATCase variants containing circularly permuted c chains
Differential scanning microcalorimetry was used to determine the effects of the circular permutation on the thermal stability of the holoenzymes. As shown in Figure 5 ▶, ATCaseH6 unfolded as a single, slightly asymmetric endotherm with a transition midpoint (Tm) at 63.5°C. The wild-type enzyme unfolded at a slightly higher temperature, 64.1°C (Table 3). Thermograms for two permuted variants, I215/E217-VR and Q137/T136, are shown as examples of holoenzymes exhibiting slightly and substantially reduced melting temperatures (Fig. 5 ▶). The melting temperatures and values of ΔTm, representing the change in melting temperature relative to ATCaseH6, are summarized in Table 3. Most of the variants exhibited slightly reduced transition temperatures, with ΔTm values ranging from −0.7°C for V169/S171-LR to −4.1°C for R56/R54-IR. However, two variants, both with c chains beginning at Q137, showed markedly reduced melting temperatures, almost 14°C lower than ATCaseH6. It is noteworthy that the Tm value (59.8°C) of the Q137/S131 variant, which has a 5-residue deletion in the permuted c chains, is only slightly decreased.
Fig. 5.

Thermograms of ATCase variants obtained by differential scanning microcalorimetry. Holoenzymes containing circularly permuted c chains were scanned at 15°C/h as described in Materials and Methods. ATCaseH6 (solid line), I215/E217-VR (broken line), and Q137/T136-HKVN (dotted line) are shown.
Discussion
Changes in functional properties of ATCase resulting from circular permutation of c chains
The initial experiments on ATCase containing circularly permuted c chains generated by site-specific techniques demonstrated that a variant with c chains beginning at D236, which is in a flexible loop, was catalytically active but devoid of allosteric properties (Yang and Schachman 1993). In subsequent studies with other ATCase permutants having termini located in other flexible loops, it was shown that the enzymes were both catalytically active and allosteric with only minor changes in kinetic constants and Hill coefficients (Zhang and Schachman 1996). In contrast, one construct that would have placed the termini at M104 in a β strand in the interior of the protein failed to express detectable levels of enzyme activity or holoenzyme. Another permutant having termini at M227 in a different β strand was found to be produced in large amounts but was only partially active (Zhang and Schachman 1996). Based on these observations, it is clear that the locations of the newly introduced termini can have different effects on the folding of the polypeptide chains, assembly into stable holoenzymes, and their catalytic and allosteric properties. The method of random circular permutation (Graf and Schachman 1996) was devised in part to address these issues and generate a library of ATCase molecules with diverse properties.
The principal advantage of the random permutation method is that the selection and screen permit the identification of potential clones leading to enzyme activity or reasonably stable holoenzymes with structures comparable to wild-type ATCase. From the selected clones, variants whose termini are located in elements of secondary structure are easily identified. Of the nine purified variants having termini within or adjacent to helices, only I215/E217-VR and A32/A32-NKVN possessed considerable enzyme activity. These data, taken together with results of previous studies (Zhang and Schachman 1996), indicate that placing the termini in elements of secondary structure often leads to major changes in tertiary structure or active site geometry. However, it should be noted that the library of variants investigated thus far is only a small fraction of those potentially available and that the topological map in Figure 1 ▶ is only partially saturated. The observation that some helices can be interrupted without the loss of enzyme activity justifies further investigations aimed at determining whether active enzyme can be produced in variants whose ends are located in β strands as well.
On the basis of site-specific mutagenesis experiments which have demonstrated that amino acid substitutions at residue Q137 (Stebbins et al. 1989) and at other positions lead to a substantial reduction in Vmax and increase in K0.5 for aspartate, it is not surprising that circular permutation of the c chains at positions 137, 169, and 170 have major effects on catalysis. Similarly, disruption of the polypeptide chain at R54 and R56 likely causes conformational changes affecting the phosphate-binding loop comprising residues (52–55) which are known to be implicated in the binding of carbamoyl phosphate (Stevens et al. 1991).
Effect of disruption of α helices on stability
Previous studies of the effects of amino acid deletions at the C termini of the c chains in wild-type ATCase demonstrated that removal of the last five residues had little impact on the enzyme activity or stability of the holoenzyme. However, chain-termination mutations causing truncation of the chains at several positions within the large interdomain helix (Helix 12) resulted in pyrimidine auxotrophy, indicating that most of this helix is essential for proper folding and assembly of a stable enzyme (Peterson and Schachman 1991). Therefore, it was of interest to determine the effect of disruption of other helices on the assembly and stability of ATCase. Based on results with the three permuted variants having the N terminus at Q137 in the 15-residue interdomain helix (Helix 5), that secondary structural element can tolerate interruptions, deletions, and insertions without impairing drastically the in vivo folding of the chains, as well as assembly and stability of the holoenzyme. It is particularly striking that the variant, Q137/S131, with the 5-residue deletion has a higher Tm than the other variants having no deletions or insertions (Table 3). In addition, Helices 6 and 8 can undergo breaks with minimal effects on protein expression and stability. It is unclear whether disruption of the remaining helices would have a deleterious effect on the folding or association of c chains and on enzyme activity.
Circularly permuted T4 lysozyme having termini within a large interdomain helix has been shown to be less stable than its wild-type counterpart (Llinás and Marqusee 1998). Despite this destabilization, the lysozyme variant exhibited ∼10% of wild-type activity, suggesting that the permuted protein was similar to the native protein in tertiary structure. Although this variant was grossly destabilized, it is possible that interruptions in other helices of T4 lysozyme may be tolerated without substantial impact on stability.
Structure of ATCase-containing permuted c chains
Several questions regarding the three-dimensional structure of permuted variants of ATCase are crucial in understanding effects of relocation of the chain termini. First, if new chain termini are located in a hydrophobic region of the native protein, do they assume a similar position in the permuted protein, or do the ends alter their position so that they are in an energetically favored region? Can the oppositely charged —NH3+ and —COO− groups neutralize each other via ionic interactions? Another important question is whether interrupted helices produced by circular permutation fold into two shorter helices, thereby maintaining many of the tertiary structural interactions. Answering these questions requires crystallographic studies of variants such as the three ATCase permutants with c chains beginning at residue Q137. The two variants Q137/T136-HKVN and Q137/T136, each of which has Met at the N terminus, are significantly less stable than the variant Q137/S131, which also has Met at the N terminus but has a deletion of five residues. Further investigations will doubtless yield invaluable information on whether insertions of additional residues, along with the introduction of ion pairs, are more destabilizing than deletions that do not cause packing problems.
Effects on N-terminal processing
As observed in previous permutation studies of ATCase catalytic chains (Yang and Schachman 1993; Zhang and Schachman 1996), the locations of the new termini had a large effect on N-terminal processing. The degree of processing is thought to be a function of the second residue (Hirel et al. 1989). However, for some circularly permuted proteins, the degree of N-terminal processing differs significantly from the prediction based on the sequence (Zhang and Schachman 1996). One additional permuted c-chain variant (V169/S171-LR) exhibits substantially less processing than that based on the sequence. Altogether, about one-third of the permuted c-chain variants analyzed thus far have processing that differs from predictions. We surmise that another factor determines the degree of processing, namely, the location of the N terminus in the three-dimensional structure. This would be reasonable if protein folding were much faster than recognition by an N-terminal transferase, which is likely in most cases. The approach using circular permutation may provide more insight into the analysis of N-terminal processing in terms of the location of the N terminus in the three-dimensional structure.
Materials and methods
Materials
Vent DNA polymerase and T4 DNA ligase were from New England Biolabs. DNA Polymerase I (Klenow Fragment), Calf Intestinal Phosphatase, and Q-Sepharose Fast Flow resin were from Pharmacia Biotech (Uppsala, Sweden). Ni-NTA agarose was from Qiagen, Inc. 14C-CbmP was from American Radiolabeled Chemicals. PALA (lot MK45-89-1) was kindly provided by Dr. Robert R. Engle (Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute, National Institutes of Health, Bethesda, MD).
Methods
Circular permutation
Random circular permutation of the Escherichia coli pyrB gene, encoding the catalytic chains of ATCase, was performed as reported (Graf and Schachman 1996). The resulting library was transformed into E. coli strain HS533, which lacks the chromosomal copy of pyrB. Clones producing active ATCase were selected by complementation in this ATCase-deficient strain, and those producing ATCase in substantial quantity were identified using an immunoblot screen or by nondenaturing PAGE. The DNA sequence of clones producing active or stable enzyme was determined to deduce the protein sequence. The permutant Q137/T136 was constructed by site-directed mutagenesis using PCR techniques (Zhang and Schachman 1996).
Protein expression and purification
ATCasewt was purified by the method of Gerhart and Holoubek (1967) except that the anion-exchange resin Q-Sepharose (Pharmacia Biotech, Uppsala, Sweden) was substituted for DEAE-Sephadex. Purification was facilitated by subcloning the permuted genes into a plasmid containing a modified pyrI gene encoding a hexa-His sequence at the N terminus of the regulatory chains. ATCaseH6 and the variants containing circularly permuted c chains were expressed in strain HS533 and purified as described (Graf and Schachman 1996).
Mass spectrometry
Prior to mass spectrometry the c and His-tagged regulatory chains of the variants were separated by reversed-phase HPLC on a Vydac C18 column. Masses were determined on a Hewlett Packard S989A electrospray ionization mass spectrometer.
Enzyme kinetics
Steady-state kinetic assays were performed with 14C-CbmP as described (Davies et al. 1970). Assays were performed at 30°C in reactions containing 50 mM MOPS • KOH at pH 7.0, 0.2 mM EDTA, 2 mM βME, and 4 mM CbmP. Enzyme concentrations were 1–3 μg/mL and aspartate concentrations 0–30 mM. Circularly permuted variants that displayed greatly reduced catalytic activity were assayed at 30–600 μg/mL and were estimated to exhibit Vmax < 0.01 mmole carbamoyl aspartate/mg per hour (<0.1% of the wild-type value).
Ultraviolet difference spectroscopy
Ultraviolet absorption difference spectra in the absence and presence of the bisubstrate analog PALA (Collins and Stark 1971) were measured using a Cary 3 spectrophotometer. Spectra were recorded from 310 to 275 nm at 0.2-nm intervals, using 1-cm path length cuvettes. Difference spectra between the unliganded and liganded enzymes, obtained following addition of PALA to 4 and 40 PALA/active site, showed a characteristic minimum at 285 nm and a maximum at 290 nm. Results are recorded as the change in absorbance, ΔA290–285, expressed in units of per centimeter per milligram per milliliter.
Analytical ultracentrifugation
Sedimentation velocity experiments at protein concentrations 2.5 mg/mL were performed with a Beckman XL-A analytical ultracentrifuge equipped with absorption optics. Samples contained no PALA or 0.18-mM PALA (4 PALA/active site). Sedimentation coefficients were determined by using the second moment or the time derivative method provided in the XLA-Velocity software (Beckman, Inc.). In each experiment, the different cells contained solutions of unliganded enzyme and PALA-liganded enzyme (about 4 PALA/active site). The reported sedimentation coefficients and values of Δs/s were not corrected for density or viscosity of the solvent or for the change in buoyant weight caused by bound ligands.
Differential scanning calorimetry
Thermal stabilities of the variants were measured calorimetrically with proteins dialyzed against 40 mM KBO3, 0.2 mM EDTA at a scan rate of 15°C/h as described (Peterson and Schachman 1991). A VP-DSC calorimeter (MicroCal, Inc.) was employed using protein concentration 0.4 mg/mL. At least two scans were done using independent protein preparations.
Acknowledgments
This research was supported by National Institute of General Medical Sciences research grant GM 12159 to H.K. Schachman, National Research Service Award 19014 to P.T. Beernink, and a fellowship to R. Graf from the Swiss National Science Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
Asp, aspartate
ATCase, aspartate transcarbamoylase
βME, β-mercaptoethanol
c, catalytic polypeptide chain
CbmP, carbamoyl phosphate
DSC, differential scanning calorimetry
EDTA, ethylenediaminetetraacetic acid
IPTG, isopropyl β-d-thiogalactopyranoside
K0.5 (Asp), apparent dissociation constant for aspartate
Met, methionine
MOPS, 3-(N-morpholino)propanesulfonic acid
OAc, acetate
PAGE, polyacrylamide gel electrophoresis
PALA, N-(phosphonacetyl)-l-aspartate
r, regulatory polypeptide chain
H6 as subscript, hexa-His sequence at the N terminus of the regulatory chain
wt as subscript, wild type
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.39001.
References
- Buchwalder, A., Szadkowski, H., and Kirschner, K. 1992. A fully active variant of dihydrofolate reductase with a circularly permuted sequence. Biochemistry 31 1621–1630. [DOI] [PubMed] [Google Scholar]
- Chu, V., Freitag, S., Le Trong, I., Stenkamp, R., and Stayton, P. 1998. Thermodynamic and structural consequences of flexible loop deletion by circular permutation in the streptavidin–biotin system. Protein Sci. 7 848–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, K.D. and Stark, G.R. 1971. Aspartate transcarbamylase. Interaction with the transition state analogue N-(phosphonacetyl)-l-aspartate. J. Biol. Chem. 246 6599–6605. [PubMed] [Google Scholar]
- Davies, G.E., Vanaman, T.C., and Stark, G.R. 1970. Aspartate transcarbamylase. Stereospecific restrictions on the binding site for l-aspartate. J. Biol. Chem. 245 1175–1179. [PubMed] [Google Scholar]
- Garrett, J.B., Mullins, L.S., and Raushel, F.M. 1996. Are turns required for the folding of ribonuclease T1? Protein Sci. 5 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart, J.C. and Holoubek, H. 1967. The purification of aspartate transcarbamylase of Escherichia coli and separation of its protein subunits. J. Biol. Chem. 242 2886–2892. [PubMed] [Google Scholar]
- Goldenberg, D.P. and Creighton, T.E. 1983. Circular and circularly permuted forms of bovine pancreatic trypsin inhibitor. J. Mol. Biol. 165 407–413. [DOI] [PubMed] [Google Scholar]
- Graf, R. and Schachman, H.K. 1996. Random circular permutation of genes and expressed polypeptide chains: Application of the method to the catalytic chains of aspartate transcarbamoylase. Proc. Natl. Acad. Sci. USA 93 11591–11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, M., Piotukh, K., Borriss, R., and Heinemann, U. 1994. Native-like in vivo folding of a circularly permuted jellyroll protein shown by crystal structure analysis. Proc. Natl. Acad. Sci. USA 91 10417–10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann, U. and Hahn, M. 1995. Circular permutations of protein sequence: Not so rare? Trends Biochem. Sci. 20 349–350. [DOI] [PubMed] [Google Scholar]
- Hennecke, J., Sebbel, P., and Glockshuber, R. 1999. Random circular permutation of DsbA reveals segments that are essential for protein folding and stability. J. Mol. Biol. 286 1197–1215. [DOI] [PubMed] [Google Scholar]
- Hirel, P.H., Schmitter, M.J., Dessen, P., Fayat, G., and Blanquet, S. 1989. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc. Natl. Acad. Sci. USA 86 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett, G.J. and Schachman, H.K. 1977. Allosteric regulation of aspartate transcarbamoylase. Changes in the sedimentation coefficient promoted by the bisubstrate analogue N-(phosphonacetyl)-l-aspartate. Biochemistry 16 5077–5083. [DOI] [PubMed] [Google Scholar]
- Johnson, J.L. and Raushel, F.M. 1996. Influence of primary sequence transpositions on the folding pathways of ribonuclease T1. Biochemistry 35 10223–10233. [DOI] [PubMed] [Google Scholar]
- Ke, H., Lipscomb, W.N., Cho, Y., and Honzatko, R.B. 1988. Complex of N-phosphonacetyl-l-aspartate with aspartate carbamoyltransferase. X-ray refinement, analysis of conformational changes and catalytic and allosteric mechanisms. J. Mol. Biol. 204 725–747. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. Molscript: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Lipscomb, W.N. 1994. Aspartate transcarbamylase from Escherichia coli: Activity and regulation. Adv. Enzymol. 68 67–151. [DOI] [PubMed] [Google Scholar]
- Llinás, M. and Marqusee, S. 1998. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 7 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger, K., Hommel, U., Herold, M., Hofsteenge, J., and Kirschner, K. 1989. Correct folding of circularly permuted variants of a βα barrel enzyme in vivo. Science 243 206–210. [DOI] [PubMed] [Google Scholar]
- Mullins, L.S., Wesseling, K., Kuo, J.M., Garrett, J.B., and Raushel, F.M. 1994. Transposition of protein sequences: Circular permutation of ribonuclease T1. J. Am. Chem. Soc. 116 5529–5533. [Google Scholar]
- Nakamura, T. and Iwakura, M. 1999. Circular permutation analysis as a method for distinction of functional elements in the M20 loop of Escherichia coli dihydrofolate reductase. J. Biol. Chem. 274 19041–19047. [DOI] [PubMed] [Google Scholar]
- Otzen, D. and Fersht, A. 1998. Folding of circular and permuted chymotrypsin inhibitor 2: Retention of the folding nucleus. Biochemistry 37 8139–8146. [DOI] [PubMed] [Google Scholar]
- Peterson, C.B. and Schachman, H.K. 1991. Role of a carboxyl-terminal helix in the assembly, interchain interactions, and stability of aspartate transcarbamoylase. Proc. Natl. Acad. Sci. USA 88 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper, U., Hayakawa, K., Li, Z., and Herzberg, O. 1997. Circularly permuted β-lactamase from Staphylococcus aureus PC1. Biochemistry 36 8767–8774. [DOI] [PubMed] [Google Scholar]
- Stebbins, J.W., Xu, W., and Kantrowitz, E.R. 1989. Three residues involved in binding and catalysis in the carbamyl phosphate binding site of Escherichia coli aspartate transcarbamylase. Biochemistry 28 2592–2600. [DOI] [PubMed] [Google Scholar]
- Stevens, R.C., Chook, Y.M., Cho, C.Y., Lipscomb, W.N., and Kantrowitz, E.R. 1991. Escherichia coli aspartate carbamoyltransferase: The probing of crystal structure analysis via site-specific mutagenesis. Protein Eng. 4 391–408. [DOI] [PubMed] [Google Scholar]
- Tsuji, T., Yoshida, K., Satoh, A., Kohno, T., Kobayashi, K., and Yanagawa, H. 1999. Foldability of barnase mutants obtained by permutation of modules or secondary structure units. J. Mol. Biol. 286 1581–1596. [DOI] [PubMed] [Google Scholar]
- Uversky, V., Kutyshenko, V., Protasova, N., Rogov, V., Vassilenko, K., and Gudkov, A. 1996. Circularly permuted dihydrofolate reductase possesses all the properties of the molten globule state, but can resume functional tertiary structure by interaction with its ligands. Protein Sci. 5 1844–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera, A.R., Blanco, F.J., and Serrano, L. 1995. The order of secondary structure elements does not determine the structure of a protein but does affect its folding kinetics. J. Mol. Biol. 247 670–681. [DOI] [PubMed] [Google Scholar]
- Viguera, A., Serrano, L., and Wilmanns, M. 1996. Different folding transition states may result in the same native structure. Nat. Struct. Biol. 3 874–880. [DOI] [PubMed] [Google Scholar]
- Wieligmann, K., Norledge, B., Jaenicke, R., and Mayr, E.M. 1998. Eye lens βB2-crystallin: Circular permutation does not influence the oligomerization state but enhances the conformational stability. J. Mol. Biol. 280 721–729. [DOI] [PubMed] [Google Scholar]
- Yang, Y.R. and Schachman, H.K. 1993. Aspartate transcarbamoylase containing circularly permuted catalytic polypeptide chains. Proc. Natl. Acad. Sci. USA 90 11980–11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P. and Schachman, H.K. 1996. In vivo formation of allosteric aspartate transcarbamoylase containing circularly permuted catalytic polypeptide chains: Implications for protein folding and assembly. Protein Sci. 5 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T., Bertelsen, E., Benvegnu, D., and Alber, T. 1993. Circular permutation of T4 lysozyme. Biochemistry 32 12311–12318. [DOI] [PubMed] [Google Scholar]