

Abstract
The phenomenon of entropy–enthalpy (S-H) compensation is widely invoked as an explanatory principle in thermodynamic analyses of proteins, ligands, and nucleic acids. It has been suggested that this compensation is an intrinsic property of either complex, fluctuating, or aqueous systems. The questions examined here are whether the observed compensation is extra-thermodynamic (i.e., reflects anything more than the well-known laws of statistical thermodynamics) and if so, what does it reveal about the system? Compensation is rather variably defined in the literature and different usages are discussed. The most precise and interesting one, which is considered here, is a linear relationship between ΔH and ΔS for some series of perturbations or changes in experimental variable. Some recent thermodynamic data on proteins purporting to show compensation is analyzed and shown to be better explained by other causes. A general statistical mechanical model of a complex system is analyzed to explore whether and under what conditions extra-thermodynamic compensation can occur and what it reveals about the system. This model shows that the most likely behavior to be seen is linear S-H compensation over a rather limited range of perturbations with a compensation temperature Tc = dΔH/dΔS within 20% of the experimental temperature. This behavior is insensitive to the details of the model, thus revealing little extra-thermodynamic or causal information about the system. In addition, it will likely be difficult to distinguish this from more trivial forms of compensation in real experimental systems.
Keywords: Entropy, enthalpy compensation, protein thermodynamics
The phenomenon of entropy–enthalpy compensation (referred to hereafter as compensation) is widely invoked as an explanatory principle in thermodynamic analyses of proteins, ligands, and nucleic acids. A far-from-exhaustive literature search in biological and chemical databases using the keywords entropy, enthalpy, and compensation yields >200 references to date. The term entropy-enthalpy compensation, however, is applied variably in this literature. The phenomena described by this term can be grouped into four categories, as follows:
- The concomitant increase in S and H with temperature, basically, a restatement of the thermodynamic definitions
where H, S, and T are the enthalpy, entropy, and absolute temperatures, respectively, and the derivatives are taken at constant pressure. Depending on whether Cp is temperature dependent or not and the range of temperatures examined, a plot of H versus S for a series of experiments at different temperatures may appear linear.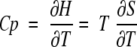
For some series of perturbations or changes in experimental variable other than temperature, ΔS and ΔH have the same sign. This is referred to here as the weak form of compensation. Examination of the statistical mechanical definitions of S and H in terms of the partition function (e.g., Hill 1986) shows that they depend in the same qualitative way on the distribution of the system among different energy levels. Preferential distribution into the lower energy states will lower the mean energy (enthalpy) and lower the entropy too. This also is an expression of well-known laws of statistical thermodynamics and reflects the fundamental importance of free energy G = H − TS in describing the behavior of the system.
A linear correlation between ΔS and ΔH of some process for a series of homologous compounds, a series of perturbations of the molecules involved, or some other regular variation of experimental conditions (other than temperature). This is referred to here as the strong form of S-H compensation. One can further classify its application in the literature to two kinds of experiments, as follows: (a) The first kind is those involving a given experimental measurement on a homologous series of compounds. Examples include the solvation thermodynamics of a solute series or the binding thermodynamics of a ligand series, in which the variation is due to the number or size or similar substituents. To quote Lumry (1995), "Linear compensation behavior is often a manifestation of the presence of a source of additivity. . . . (It) is deceptive in promising more than it delivers. In fact its profundity goes no further than indicating that the members of a series share a single source of additivity." (b) The second kind is situations in which a priori the range of ΔGs that can be observed is small. Linear correlation between ΔS and ΔH follows immediately from ΔH − TΔS = ΔG ≈ Constant. Small is defined here with respect to the range of observed ΔH values. The reasons for the narrow range of ΔGs may be biological (evolutionary) or experimental.
Examples of compensation that fall into these three categories are defined as secondary, in that they are better explained either in terms of well-known thermodynamic laws (1, 2) or by which systems were selected and which measurements were made (3). In contrast, extra-thermodynamic compensation is defined here as a linear relationship between ΔH and ΔS, which does not fall into the third category above. The key quantity provided by such a relationship is the slope, which defines a compensation temperature Tc = dΔH/dΔS. The implication (or hope) is that for extra-thermodynamic compensation, Tc should reveal some mechanistic or extra-thermodynamic information about the system that cannot be a priori deduced from the laws of statistical thermodynamics. Examples might be information about the shape of the potential energy surface, the distribution of energy levels available to the system, or the interaction between different components of the system.
Discussion
In most thermodynamic experiments only ΔG and ΔH are measured independently, ΔS being obtained by subtraction. If |ΔG| < |ΔH|, which is very often the case, then the high correlation between errors in ΔH and ΔS can produce linear ΔS-ΔH plots with a high correlation coefficient. This possibility was originally discussed by Lumry and Rajender (1970) in a detailed review of compensation. A concise discussion of this was presented by Krug et al. (1976), who showed that a simple statistical test can be used to determine the significance of such ΔS versus ΔH plots. The confidence interval for Tc is determined from a linear regression analysis of this plot and we ask whether the experimental temperature T (or harmonic mean experimental temperature if data are obtained at different temperatures) lies outside this confidence interval. Thus, the correlation would not be significant at the 95% confidence level if
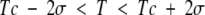 |
1 |
where σ is the estimated standard error in Tc from the fit. Krug et al. (1976) used this test to examine 38 data sets purportedly showing high ΔS-ΔH compensation. For only three of these, according to Krug et al. (1976), "was the hypothesis rejected that the observed compensation pattern can be explained as an artifact." Unfortunately, the simple test proposed in this paper does not appear to be widely known or applied, perhaps because of the predominantly negative results it produced. Below I reexamine three recent sets of protein thermodynamic data using this test.
Calcium binding to proteins
Linear ΔS versus ΔH plots obtained from calcium-binding proteins by Kuroki et al. (1992) has been cited as a particularly significant example of S-H compensation by Qian and Hopfield (1996).
Small globular protein unfolding
Privalov and Gill (1988) measured the unfolding free energy and enthalpy of several small proteins using calorimetry, and they found that ΔH and ΔS of unfolding on a per-residue basis are highly correlated.
Unfolding of cytochrome c
Milne et al. (1999) measured the hydrogen-exchange protection factors for amide proteins in oxidized and reduced cytochrome c at various temperatures. From the T dependence of these protection factors, they determined ΔS and ΔH for opening at each amide and found a high correlation. They found no reason for this correlation, involving, as it does, a mixture of local protein fluctuations and larger unfolding events at different parts of the protein.
ΔS versus ΔH plots for these three experiments are shown by the solid symbols in Figure 1A ▶–C, respectively. In each case, an impressive linear plot resulted, with a high correlation coefficient (Table 1, Col. 2). This table also shows, however, that for the first two experiments T falls within the 95% confidence limits of Tc (Col. 4), whereas for the third set, T falls just outside the confidence limits. Thus, in spite of the linearity, the S-H correlations range from clearly not significant to barely significant. To show these conclusions in a more graphic and forceful way, I regenerated each ΔS versus ΔH plot using the actual ΔG data combined with random ΔH values spanning about the same range. The randomly selected ΔHs give equally good or slightly better fits with a statistically indistinguishable slopes (Fig. 1A ▶–C, open symbols; Table 1).
Fig. 1.

Entropy (expressed as TS at 298K)–enthalpy compensation plots for (A) calcium binding to proteins, (B) protein unfolding, (C) hydrogen exchange in cytochrome c, and (D) alkane solvation. Using experimental ΔG and ΔH data taken from Kuroki et al. (1992), Privalov and Gill (1988), Milne et al. (1999), and Ben-Naim and Marcus (1984), respectively (▪). Using randomly generated ΔH values with experimental ΔG's (○).
Table 1.
Summary of experimental ΔS–ΔH data
| Data | R2 (Expt. ΔH) | T (K) | Tc (K) | R2 (Random ΔH) | ΔG range (kcal/mole) |
| Calcium binding | 0.960 | 298 | 250–310 | 0.976 | −9 ± 2 |
| Protein unfolding | 0.983 | 298 | 263–311 | 0.984 | 0.08 ± .02 |
| Hydrogen exchange | 0.970 | 293 | 251–283 | 0.974 | 9 ± 2 |
| Alkane vaporization | 0.966 | 298 | 157–169 | 0.750 | 7 ± 2.5 |
Lest the impression arise that these two tests would yield a negative result for any set of ΔS-ΔH data, data for entropy and enthalpy of solvation of the linear alkane series pentane through hexadecane (Ben-Naim and Marcus 1984) are presented in Figure 1D ▶ and Table 1. Here, T falls many standard deviations outside the Tc confidence limits, and the random ΔHs produce a clearly inferior fit with a significantly different slope (Tc = 330K). However, this is simply an example of additivity producing a linear ΔS-ΔH plot (category 3a above).
In each of the three protein data sets, reasonable arguments can be made that prior experimental or biological factors constrain the range of ΔGs that can be observed. For calcium-binding proteins, the range of affinities must lie within some biologically functional window determined by in vivo calcium levels, a requirement to modulate activity by removing calcium and such. For small protein unfolding, ΔG must have some minimum value (<6 kcal/mole) to have any stability, whereas the requirement for equilibrium reversible calorimetry in aqueous solution puts an upper bound on the stability of proteins for which one can easily obtain measurements. Thus, the contribution to stability per residue for easily measurable proteins is constrained to be a few tenths of a kcal/mole. For hydrogen exchange data on cytochrome c, the maximum protection factor has an upper bound given by the protein's global stability (≈13 kcal/mole for the oxidized form), whereas the difficulty of measuring rapidly exchanging hydrogens puts a lower bound on the measurable free energy values (≈6 kcal/mole). Thus for all three sets of experimental data the a priori range of observable ΔGs is small compared to the average values (Table 1, Col. 6) or the average enthalpy. Other examples of this no doubt can be found in the literature. For example, another recent study of hydrogen exchange in lysozyme by Dixon et al. (2000) also produced an impressive ΔS versus ΔH plot, which was attributed to an even more narrow observation window for ΔG (a range of only ≈2.3 kcal/mole). In contrast, Gallicchio et al. (1998) provide examples of non-compensation, where S and H change linearly but with opposite sign.
In each of the three protein data sets, it is statistically likely that the compensation is produced by the high correlation between the errors in estimating H and S. One cannot, however, rule out the possibility that compensation with Tc = T would be seen in these systems if the H and S were measured more precisely, but on balance, nothing in the data examined so far provides convincing evidence of extra-thermodynamic compensation.
It has been suggested that compensation is an intrinsic property of complex systems that have many soft modes of fluctuation, which would include aqueous solutions and soluble proteins (Lumry and Rajender 1970; Weber 1995; Qian and Hopfield 1996; Qian 1998). Below I examine this using a simple statistical mechanical model of a complex system to explore whether and under what conditions extra-thermodynamic S-H compensation can occur, and what it can tell us about the system. For this purpose, I adopt the following working description of a complex system: It is composed of many atoms, and hence many degrees of freedom, governed by some potential energy function (Hamiltonian). This Hamiltonian includes many kinds of interactions (van der Waals, electrostatic, torsions, etc.). These interactions have different functional forms and distance dependencies, which result in a complex multidimensional energy surface with a very large number of closely spaced minima (energy states). Let the number of states with an energy within some small range U to U + δU be ω(U)δU, where ω(U) is the density of states. The configurational part of the partition function Q is obtained by integrating overall energy levels
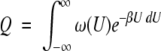 |
2 |
where β = 1/kT, T is the temperature and k is the Boltzmann constant. To model the effect of a perturbation, it is assumed that the energy levels in the range U′ to U′ + δU are significantly perturbed by raising them by an amount ΔU. The partition function of the perturbed system Q′ can be written in terms of the unperturbed system as
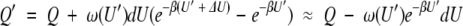 |
3 |
where the second equality here defines a significant perturbation to mean ΔU > 3kT, so the first exponential term is small compared to the second. The change in free energy produced by the perturbation is then given by
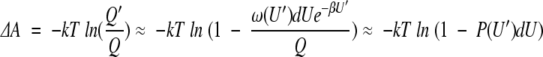 |
4 |
where P(U′) = ω(U′)e−βU′ is the unperturbed probability distribution, that is, the probability of finding the system in a state with energy U′ in the unperturbed system. In a complex system with many closely spaced energy levels, the probability of being in any small range of energy levels is small, that is, P(U′)dU << 1, and a linear expansion of the logarthmic term gives
 |
5 |
The mean energy in the unperturbed system is
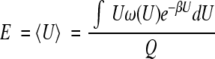 |
6 |
(The difference between mean energy E and enthalpy H in most biochemical experiments is small and no distinction is made between them here.) With the above definition of a significant perturbation, the mean energy in the perturbed system can be written as
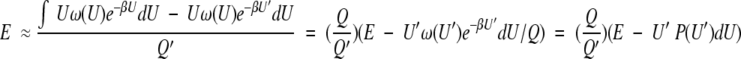 |
7 |
Using equation 3 to substitute for Q′, the ratio of the partition functions in equation 5 may be written
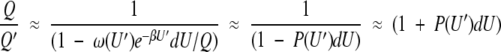 |
8 |
The last equality again uses the fact that P(U′)dU << 1, allowing a linear expansion of the reciprocal. Substituting equation 7 into equation 6 and subtracting the energy of the unperturbed system gives the change in mean energy
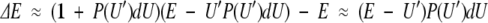 |
9 |
In the last equality the quadratic term U′(P(U′)dU)2 has been dropped because it is negligible compared to the linear terms. Finally, an expression for the entropy change may be obtained using TS = E − A:
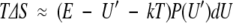 |
10 |
It should be noted that equations 5, 9, and 10 are general. They do not depend on the particular distribution of states, providing the following two assumptions are satified: (1) A small number of the states are perturbed and (2) the size of the perturbation is significant. Moreover, the magnitude of the perturbation in A, E, and S depends on the initial energy of the perturbed state(s) and how many are perturbed but not on how much the states are perturbed. These equations also apply to the situation in which the perturbation results in a lowering of the energy levels simply by exchanging the role of perturbed and unperturbed states. This results in free energy, entropy, and enthalpy changes of equal magnitude and opposite sign. In this case U′ refers to the final energy of the perturbed state(s), rather than the initial energy.
One may define a compensation temperature in this model by
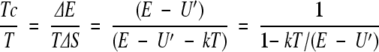 |
11 |
Clearly this is not a constant, but it depends on where the energy of the perturbed states lies with respect to the mean energy.
To provide a concrete example of the entropy–enthalpy behavior in this model, it is useful to use some specific distribution of energy levels, but it should be stressed that the general conclusions do not depend on the specific form. Given the high dimensionality and complexity of the energy surface of a protein it seems reasonable that as one changes any given conformational degree of freedom, there are relatively few minima that can be accessed that are either very low or very high in energy. Most will cluster around some mean value. In particular I will assume a Gaussian density of such minima ω(U) of mean Uo, width σ permitting an analytical treatment. Because the statistical mechanical behavior is independent of the zero point of the energy scale, the presentation can be further simplified, without loss of generality, by setting Uo = 0.
It also seems reasonable that because many competing interactions from many different atoms contribute to the energy, there is, on average, little correlation in the change produced by simultaneously perturbing any two degrees of freedom. If the first produces an increase in energy, the second is, on average, as likely to decrease the energy as increase it. In other words, each degree of freedom can be treated independently and the total configurational partition function Q can be approximated as the product of the partition functions for each degree of freedom Q = ⇁qj, and equations 5, 9, and 10 will apply to each qj. I will consider below whether correlations between different degrees of freedom affect the conclusion to be drawn from this model.
For a Gaussian density of states, the configurational part of the partition function for a given degree of freedom qj is
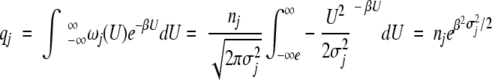 |
12 |
where β = 1/kT, and nj is the total number of states accessible to the jth degree of freedom The probability distribution in the unperturbed system is (see Fig. 2A ▶)
Fig. 2.

(A) Density of states (□) and occupancy of those states at 298K (▪) as a function of energy in the Gaussian density of states model. (B) Change in mean energy E (□) and entropy TS (▴) at 298K as a function of the energy of the perturbed states.
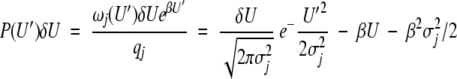 |
13 |
Using this probability distribution in equations 9 and 10, a plot of ΔS versus ΔE can be generated for a systematic series of perturbations, corresponding to a series of experimental manipulations that affect different energy levels, by varying U′. The result is shown in Figure 3 ▶. The resulting plot is ellipsoidal with the major axis aligned along the Tc = T direction. To check the validity of the mathematical approximations made in the derivation of equations 9 and 10, the exact partition function for the perturbed system (equation 3) was evaluted numerically for the Gaussian model. The same values of dE and dS, to within numerical precision, were obtained as from the approximate analytical results, equations 9 and 10.
Fig. 3.

Entropy (TS) vs. energy (E) plot for the Gaussian density of states model at 298K, with energy level spreads (sd, σ) of 0.5 (x), 1 (□), 2 (♦), and 3 (▪) kcal/mole, respectively.
The ellipsoidal profile can be explained with reference to equations 9 and 10 and Figure 2A ▶. Consider the effect on E of a perturbation of a given set of states at U′. This is the product of the following two terms: (1) an occupancy term, P(U′)δU, which is always positive, and (2) an energy difference term, E-U′. If we perturb an energy level that is lower than the mean energy of the unperturbed system U′ < E, then ΔE is positive because on average the system spends more time in higher energy states. The reverse is true for U′ > E. For E = U′ the first term is zero, and there is no effect on the mean energy. The occupancy term is small at very low and very high energies because the probability density of these states is small, hence ΔE → 0. Thus, as increasingly higher energy states are perturbed, ΔE first increases as the occupancy term increases, drops to zero, and becomes negative as the second term changes sign, and finally returns to zero as the probability density decreases (Fig. 2B ▶). The behavior of ΔS is governed by the same equation, except E is offset by −kT. It follows the same profile: It is increasingly positive at low U′, decreasing through zero to negative values and finally returning to zero at high U′ but with a phase shift (Fig. 2B ▶). The resulting plot of ΔS versus ΔE is ellipsoidal.
What can be extracted from this ΔS-ΔE plot? The key parameter in the Gaussian density of states model is σ, the width of the density distribution, that is, the spacing of the energy levels. A more elongated ellipse indicates a greater spacing of energy levels. The direction of the major axis remains unchanged and provides no specific information. The discussion in the previous paragraph indicates that the criteria for an ellipsoidal type ΔE-ΔS plots are rather broad, requiring only that P(U′) tends to zero at high and low U′; that is, any peaked density of states distribution will produce very similar behavior.
Is such behavior seen in experimental systems? Data presented by Eftink and Biltonen (1983) for nucleotide binding to RNase show such behavior Hooked plots of ΔH versus ΔS for linked binding-conformational change equilibria are observed, which resemble portions of an ellipse. The parameter changed in these plots is the equilibrium constant for the conformational change, that is, the energy gap between the two protein conformations. Thus, their experimental situation is effectively a two-energy-level version of the model presented here. However, in any given system, it is unlikely that a sufficient range of perturbations of U′ is experimentally realizable, so complete elliptical plots are not likely to be seen. A restricted range of perturbations would effectively manifest some portion of the ellipse, as seen by Eftink and Biltonen (1983). Depending upon which portion and how much is accessed, quasi linear ΔH-ΔS plots of widely varying slope (Tc) could result. In these situations, the particular value of Tc would reveal nothing specific about energy-level distribution. Examples of non-compensation discussed by Gallicchio et al. (1998) may represent portions of the ellipse with negative slope.
The Gaussian density of state model describes the contribution from a single degree of freedom (DOF) qj. Thus, the net perturbation in ΔE or TΔS is rather small. (In fact, from equations 9 and 10, it must be <kT.) An actual experiment represents the net effect of perturbations to many DOFs. If the perturbations are not correlated, then one would expect many of the contributions to ΔE or TΔS to cancel, resulting in small net values. Thus, larger experimental ΔE or TΔS values presumably result from correlated perturbations from many degrees of freedom. In effect this would produce ΔE and ΔS values corresponding to a summation of similar elliptical curves all aligned along Tc = T. Combined with finite experimental precision, this would most likely result in a fat line with slope Tc ≈ T. This would be difficult to distinguish from the self-evident compensation of case 3. This model suggests that extra-thermodynamic compensation, if it exists, is unlikely to be observed in real experimental systems and difficult to interpret if it does.
In summary, if the range of ΔG's measured in a series of experiments is much smaller than the range of ΔH's, then with respect to ΔH, ΔG ≈ Constant. Linear dH-dS compensation follows immmediately from the relationship ΔG = ΔH − TΔS. The question then is whether this arises from (1) larger errors in determining ΔH than ΔG, (2) Some extra-experimental constraint that a priori restricts the range of observable ΔGs, or (3) some extra-thermodynamic mechanism of ΔH-ΔS compensation. For the three data sets examined here, the statistical tests strongly suggest, although they cannot prove, the first explanation. I argue that this is because extra-experimental constraints a priori restrict the range of observable ΔGs to less than the precision in dH measurements, even though the latter may be carefully measured. Nevertheless, without knowing the molecular origin of the entropy and enthalpy components and from statistical tests alone, one cannot rule out some type of extra-thermodynamic compensation of the type seen in the model presented here.
Acknowledgments
I thank Walter Englander and Bill De Grado for extensive discussions. Financial support from NIH GM 54105 and NSF MCB 9808202 is gratefully acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/
References
- Ben-Naim, A. and Marcus, Y. 1984. Solvation thermodynamics of nonionic solutes. J. Chem. Phys. 81 2016–2027. [Google Scholar]
- Dixon, M.E., Hitchens, T.K., and Bryant, R.G. 2000. Comparisons of pressure and temperature activation parameters for amide hydrogen exchange in T4 lysozyme. Biochemistry 39 248–254. [DOI] [PubMed] [Google Scholar]
- Eftink, M. and Biltonen, R. 1983. Enthalpy–entropy compensation and heat capacity changes for protein ligand interactions: General thermodynamic models and data for the binding of nucleotides to Ribonuclease A. Biochemistry 22 3884–3896. [DOI] [PubMed] [Google Scholar]
- Gallicchio, E., Kubo, M., and Levy, R.M. 1998. Entropy–enthalpy compensation in solvation and ligand binding revisited. J. Am. Chem. Soc. 120 4526–4527. [Google Scholar]
- Hill, T. 1986. An Introduction to Statistical Thermodynamics. Dover Books, New York.
- Krug, R., Hunter, W., and Grieger, R. 1976. Statistical interpretation of enthalpy–entropy compensation. Nature. 261 566–567. [Google Scholar]
- Kuroki, R., Nitta, K., and Yutani, K. 1992. Thermodynamic changes in binding of Ca2 + to a mutant human lysozyme. J. Biol. Chem. 267 24297–24301. [PubMed] [Google Scholar]
- Lumry, R. 1995. On the interpretation of data from isothermal processes. Meth. Enzymol. 259 628. [DOI] [PubMed] [Google Scholar]
- Lumry, R. and Rajender, S. 1970. Enthalpy–entropy compensation phenomena in water solutions of proteins and small molecules: A ubiquitous property of water. Biopolymers. 9 1125–1227. [DOI] [PubMed] [Google Scholar]
- Milne, J., Xu, Y., Mayne, L., and Englander, S.W. 1999. Experimental study of the protein folding landscape: Unfolding reactions in Cytochrome c. J. Mol. Biol. 290 811–822. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. and Gill, S.J. 1988. Stability of protein structure and hydrophobic interaction. Adv. Prot. Chem. 39 191–234. [DOI] [PubMed] [Google Scholar]
- Qian, H. 1998. Entropy–enthalpy compensation: Conformational fluctuation and induced fit. J. Chem. Phys. 109 10015–10017. [Google Scholar]
- Qian, H. and Hopfield, J. 1996. Entropy–enthalpy compensation: Perturbation and relaxation in thermodynamic systems. J. Phys. Chem. 105 9292–9299. [Google Scholar]
- Weber, G. 1995. Van't Hoff revisited: Enthalpy of association of protein subunits. J. Phys. Chem. 99 1052–1059. [Google Scholar]