

Abstract
A gene cluster isolated from Pseudomonas stutzeri OX1 genomic DNA and containing six ORFs codes for toluene/o-xylene-monooxygenase. The putative regulatory D subunit was expressed in Escherichia coli and purified. Its protein sequence was verified by mass spectrometry mapping and found to be identical to the sequence predicted on the basis of the DNA sequence. The surface topology of subunit D in solution was probed by limited proteolysis carried out under strictly controlled conditions using several proteases as proteolytic probes. The same experiments were carried out on the homologous P2 component of the multicomponent phenol hydroxylase from Pseudomonas putida CF600. The proteolytic fragments released from both proteins in their native state were analyzed by electrospray mass spectrometry, and the preferential cleavage sites were assessed.
The results indicated that despite the relatively high similarity between the sequences of the two proteins, some differences in the distribution of preferential proteolytic cleavages were detected, and a much higher conformational flexibility of subunit D was inferred. Moreover, automatic modeling of subunit D was attempted, based on the known three-dimensional structure of P2. Our results indicate that, at least in this case, standard modeling procedures based on automatic alignment on the structure of P2 fail to produce a model consistent with limited proteolysis experimental data. Thus, it is our opinion that reliable techniques such as limited proteolysis can be employed to test three-dimensional models and highlight problems in automatic model building.
Keywords: Monooxygenase, expression, Pseudomonas, recombinant, limited proteolysis, mass spectrometry
A gene cluster has been isolated by screening a library of Pseudomonas stutzeri OX1 genomic DNA, and cloned (Bertoni et al. 1996). Sequence analysis of the cluster revealed the presence of six open reading frames (ORFs) with a high degree of identity with components of monooxygenase systems (Bertoni et al. 1998) found in benzene- and toluene-degradative pathways. These similarities were confirmed by individual ORF subcloning, expression, and electrophoretic analyses of the expressed proteins (Bertoni et al. 1998). The results of these studies strongly suggest that the gene cluster encodes a novel oxygenase system named toluene/o-xylene-monooxygenase (Tomo). It has been shown (Bertoni et al. 1996) that the Tomo system is able to catalyze two consecutive hydroxylation reactions of toluene and o-xylene, via the intermediate production of a mixture of o-, m-, and p-cresol, and 2–3-dimethylphenol and 3–4-dimethylphenol, respectively. Moreover, Tomo is also able to degrade highly chlorinated compounds (Ryoo et al. 2000). Homology studies (Bertoni et al. 1998) suggest that P. stutzeri OX1 toluene/o-xylene-monooxygenase is a multicomponent enzyme, made up of six different subunits, similar to the composition of monooxygenases from B. cepacia AA1 (Newman and Wackett 1995), B. picketii PKO1 (Byrne et al. 1995), and P. mendocina KR1 (Yen et al. 1991; Yen and Karl 1992). In fact, each polypeptide component of Tomo shows similarities with components of several enzymatic complexes involved in the oxygenation of aromatic compounds (Bertoni et al. 1998).
The similarity between subunit D of the Tomo complex and the low molecular weight proteins TmoD from P. mendocina KR1 (Pikus et al. 1996), MmoB from M. capsulatus (bath) (Stainthorpe et al. 1990), and P2 from P. putida sp. strain CF600 (Nordlund et al. 1990) appears particularly interesting. It has been demonstrated that these proteins are involved in the stimulation of their respective hydroxylase components (Froland et al. 1992; Pikus et al. 1996; Qian et al. 1996) and do not play a direct role in the electron transport chain.
This paper reports the conformational analysis of the subunit D component of the toluene/o-xylene-monooxygenase system from P. stutzeri OX1 by the limited proteolysis/mass spectrometry approach (Zappacosta et al. 1996; Scaloni et al. 1998; Orrù et al. 1999) using several proteases as conformational probes. The same experiments were also performed on the homologous (65% similarity) P2 protein whose three-dimensional structure has been solved by NMR. The analysis indicates that P2 is very flexible, and well suited to bind and interact with the hydroxylase component of the complex.
Molecular models of subunit D were constructed by automatic modeling using the P2 protein as a template (Qian et al. 1996), and used to interpret the results of limited proteolysis/mass spectrometry. The analysis indicated that the automatically derived model is somewhat inadequate to explain all the selective cleavages found in subunit D. Our results suggest that, whereas automatic model building is a valuable system for obtaining approximate structural models, experimental validation methodologies, such as limited proteolysis, are also needed to evaluate and validate its results.
Results
Amino acid sequence of subunit D of Tomo
The primary structure of the recombinant subunit D of Tomo as derived from the translation of the coding gene sequence (Bertoni et al. 1996; Bertoni et al. 1998) was verified by the peptide mapping strategy. Aliquots of the HPLC-purified protein were digested with either cyanogen bromide or Asp-N endoprotease, and the resulting peptide mixtures were analyzed by MALDI/MS. The mass signals recorded in the spectra were mapped onto the anticipated sequence of subunit D on the basis of their mass value and the specificity of the enzyme, leading to the complete verification of the amino acid sequence of subunit D, shown in Figure 1 ▶.
Fig. 1.

Alignment of the P2 protein and Tomo D used to generate models. (#) Identical residues; (*) conservative substitutions. The sites of proteolytic cleavages (see Results section) are highlighted in red. Residues of Tomo D not aligned with the P2 sequence are shown in bold. The secondary structure of the P2 protein, determined by means of the DSSP program (Kabsch and Sander 1983), is also shown.
Topological studies of subunit D of Tomo and dmpM
The surface topology of subunit D in solution was investigated by a strategy that combines limited proteolysis experiments with mass spectrometric procedures (Zappacosta et al. 1996; Scaloni et al. 1998; Orrù et al. 1999). The overall strategy is based on the evidence that the amino acid residues located within exposed and flexible regions of the protein can be recognized by proteases, leading to a reasonably good indication of the conformation of the protein in solution.
Limited proteolysis experiments were performed by using trypsin, V-8 endoprotease, Asp-N endoprotease, chymotrypsin, elastase, and subtilisin as proteolytic probes. The protein was incubated with each protease under strictly controlled conditions to ensure the maintenance of its native conformation, and to increase the selectivity of proteases (Zappacosta et al. 1996; Orrù et al. 1999). The extent of the enzymatic hydrolysis was monitored on a time-course basis by sampling the incubation mixtures at different interval times, followed by HPLC fractionation. The fragments released from the proteins were identified by ES/MS, leading to the assignment of the cleavage sites. Preferential proteolytic sites were assigned from the identification of the two complementary peptides released from the intact protein following a single proteolytic event. When two or more sites were identified in the same experiment, these cleavages were always due to a single proteolytic event on the intact protein molecule, apparently occurring with the same kinetics of hydrolysis.
Peptides generated by subsequent digestion of larger fragments and not released from the intact protein were not considered in the interpretation of proteolysis data. As an example, Figure 2 ▶ shows the HPLC chromatograms of the aliquots withdrawn after 10 min, 30 min, and 60 min of trypsin digestion of subunit D. Under the controlled conditions used (1 : 1000 (w : w) trypsin : Tomo D), most of the protein remained undigested and only a few specific fragments were released (Fig. 2C ▶). The ES/MS analysis of individual fractions identified these fragments as the peptides 1–25, 1–56, 1–79, 80–109, and 57–109, respectively. The major component contained a mixture of the intact protein and the peptide 26–109. The presence of the complementary peptide pairs, that is, 1–25 and 26–109, 1–56 and 57–109, and 1–79 and 80–109, clearly indicates that these fragments originated from a single proteolytic event that occurred on the native protein. These data demonstrate that Arg25, Arg56 and Arg79 were the preferential tryptic cleavage sites in subunit D. Similar results were obtained when the specific proteolytic enzymes V-8 protease and endoprotease Asp-N were used.
Fig. 2.

HPLC chromatograms of the tryptic digests of subunit D of Tomo after 10 (A), 30 (B), and 60 (C) min of incubation.
Experiments were also carried out by incubating subunit D with broader specificity proteases such as chymotrypsin, elastase, and subtilisin. As an example, chymotrypsin digested subunit D preferentially at Leu8, Phe78, Phe103, and Tyr104 (Fig. 1 ▶ and Table 1) as inferred by the identification of the peptide pairs 1–8 and 9–109, 1–78 and 79–104, and the fragments 1–103 and 1–104.
Table 1.
Limited proteolysis cleavage sites in subunit Tomo D and DmpM
| Protease | Enzyme: Tomo D ratio | Sites in Tomo D | Enzyme: DmpM ratio | Sites in DmpM |
| Trypsin | 1:1000 | R25, R56, R79 | 1:100 | R39, R59 |
| Chymotrypsin | 1:500 | Y54, F78, F103, Y104 | 1:75 | W61, F84, W88 |
| Asp-N | 1:300 | D12, D28, D61 | ||
| V-8 | 1:50 | E35, E62, E82 | 1:30 | E65 |
| Subtilisin | 1:1500 | L8, Q81, S88 | 1:200 | M66, L86 |
| Elastase | 1:350 | L8, F78, F103 | 1:100 | M37, W61, W88 |
The overall data from the limited proteolysis experiments performed on subunit D are summarized in Table 1 and Figure 1 ▶.
The data indicate that the preferential proteolytic sites are scattered along the entire sequence of the protein. Besides few isolated cleavages essentially occurring within the N-terminal portion (Leu8, Asp12, and Glu35), the preferential proteolytic sites are gathered within a few well-defined regions of the subunit D sequence. Four short segments, Arg25-Asp28, Tyr54-Arg56, Asp61-Glu62 and Phe103-Tyr104, and the longer region spanning residues 78–88 were identified as accessible areas. Thus, the four short sequences might constitute mobile loops connecting ordered secondary structure elements, whereas the segment 78–88 might represent a very flexible and unstructured region.
A further consideration suggested by the limited proteolysis results concerns the three very hydrophilic regions in the sequence of subunit D (segments 38–51, 66–72, and 95–102). These segments, while containing a large number of putative cleavage sites, were resistant to proteases.
These regions were thus suspected to be either highly structured or not exposed to the solvent, although it should be noted that unpaired charged residues are rarely found buried inside proteins.
The same limited proteolysis experiments were carried out on the homologous DmpM (P2) protein for comparison. The data collected are reported in Figure 1 ▶ and Table 1, indicating that the proteolytic pattern of P2 is similar but not identical to that defined for subunit D of the Tomo complex. However, it should be emphasized that for all proteases, a higher enzyme-to-substrate ratio had to be used in the P2 experiments.
Modeling of subunit D of Tomo
The NMR structures of three different regulatory components of multicomponent oxygenases are currently available: the P2 protein, the regulatory component of a phenol hydroxylase (PDB code 1HQI), and the components of two different methane monooxygenases (PDB codes 1CKV and 2MOB). The structure of P2, the one sharing the higher sequence similarity with Tomo D, was selected as the template for the model building of Tomo D. This choice is also supported by the observation that their respective protein complexes catalyze the transformation of the same type of substrate, and it has been proposed that Tomo D and P2 act as regulatory components and are involved in substrate binding (Qian et al. 1996; Bertoni et al. 1998).
The amino acid sequences of Tomo D and P2 share 36.9% identity and 65.5% similarity in a 90-residue overlap segment. In addition, the sequence of Tomo D shows a 15-residue N-terminal extension which was not considered in the modeling procedure. The two sequences were aligned using a standard algorithm and the BLOSUM62 matrix (Fig. 1 ▶).
The structure of P2 has been solved by NMR (Qian et al. 1996), and the coordinates for 12 models are present in the Protein Data Bank entry 1HQI. P2 turned out to be extremely flexible in the experimental conditions used: the RMS deviation from the average structure of the secondary structure segments is around 2.5 Å, but the RMS deviation of these segments among the 12 different submitted models can be as high as 4.5 Å with an average value of about 3.6 Å.
The NMRCLUST algorithm (Kelley et al. 1996) was used to cluster the 12 structures into three groups. Two clusters contained only two models each, whereas eight structures gathered within the largest cluster which was selected for the modeling procedure. The remaining models were discarded, as they appeared quite different from the main group of structures.
The set of atoms whose positions are well defined throughout this reduced set of structures were then extracted and the conserved core of the selected eight models defined using the NMRCore algorithm (Kelley et al. 1997). The main portion which is consistently structurally conserved among the eight structures is composed by residues 4–11, 29–32, 34–44, and 71–73 and accounts for 34.2% of the total core. Finally, the structure closest to the centroid of the largest cluster (model 3) was selected for modeling, as it is more representative of the ensemble than the commonly used minimized average structure.
Thus, one Swiss-PDB-Viewer-project file (Guex and Peitsch 1997), containing the sequence alignment and the set of coordinates of the selected structure 3 of P2 was submitted to the automated modeling server. The resulting model was visually examined with Swiss-PDB-Viewer. As expected, given the high similarity between the template and the query sequence, the model was almost superimposable on the corresponding template structure (data not shown).
Discussion
Knowledge of a protein's tertiary structure is a prerequisite for the proper understanding of and for engineering its function. This makes the successful prediction of a protein tertiary structure a central issue in proteomics, in cases in which the experimental three-dimensional structure is not yet available. However, although prediction and modeling techniques have greatly improved in the past few years (Moult et al. 1999), simple and effective tools are needed for experimentally testing tertiary structure models. This would give confidence in the prediction results, especially when automated procedures are used to generate the models.
Limited proteolysis studies of protein molecules coupled with fast and sensitive mass spectrometry analyses of peptide fragments is one such tool. Its potency lies in the fact that proteolytic enzymes are amenable probes of protein secondary and tertiary structures (Fontana et al. 1997). Furthermore, such studies can be used both to describe surface topology and to monitor conformational changes of proteins in solution (Orrù et al. 1999)
The primary structure of Tomo D, as determined in the present study, is largely similar to that of P2 protein from P. putida, except for its 15 N-terminal amino acids (Fig. 1 ▶). The Tomo D polypeptide chain contains 109 residues, whereas P2 has only 90 residues. Both proteins were submitted to conformational analysis using the limited proteolysis/mass spectrometry approach. When the results obtained on Tomo D and P2 were compared, similar but not identical proteolytic patterns could be defined. Both proteins showed short flexible segments accessible to proteases and very likely connecting secondary structure elements and a larger loop encompassing residues 76–83 (numbers refer to Tomo D sequence). Moreover, the three hydrophilic regions resistant to proteases present in Tomo D find their homologous counterparts in P2 structure, although a low degree of sequence similarity occurs in the segment 38–51 (Fig. 1 ▶).
On the other hand, Tomo D appeared to be more flexible and/or less structured with respect to its homolog P2 as demonstrated by the lower enzyme-to-substrate ratios used in all the proteolytic experiments,. Moreover, the region encompassing residues 25–28 and 61–62 and the Glu residue at position 35, accessible to proteases in Tomo D, were not recognized in P2, although putative cleavage sites occurred at corresponding sequence positions.
Thus, despite their high degree of similarity, a number of topological differences could be mapped on the surface of the two proteins.
The results of the limited proteolysis experiments carried out on P2 were then mapped onto the three-dimensional structure of the protein (Qian et al. 1996) as a test of the reliability of our structure validation procedure. Figure 3A ▶ shows the location of the preferential proteolytic sites within model 3 of P2. The accessible segment Met54-Arg56 (numbering refers to Tomo D sequence) occurs within a flexible portion of the protein at the junction between a loop region and strand β2. Analogously, the region encompassing residues Arg76 to Met83, readily cleaved by proteases, corresponds to the long segment closing the hydrophobic cavity where the substrate is thought to bind to the protein and interact with the hydroxylase complex. It should be stressed that this segment belongs to the region showing the largest deviation of atomic coordinates among the 12 NMR structures of P2, and it has been described as highly flexible (Qian et al. 1996). Accordingly, the more representative models, as defined by the clustering algorithm, display a nonstructured region spanning residues 76–86. This finding demonstrates that sensitive conformational probes such as proteolytic enzymes were able to correctly discriminate among the ensemble of NMR structures depicted for P2.
Fig. 3.
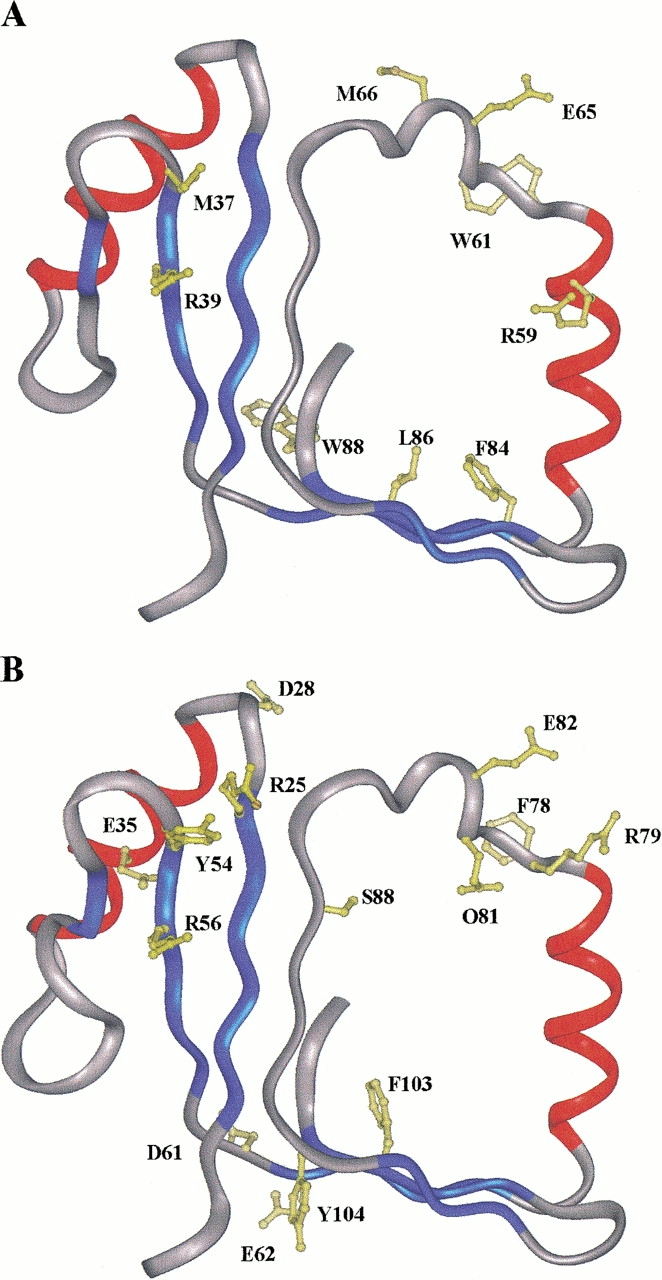
Structure 3 of the P2 template (A) and one of the modeled structures (model 3) of Tomo D (B) are shown, respectively, with the residue side chains at the limited proteolysis sites. Color codes in (C) and (D) correspond to secondary structure: α-helix (red), β-strands (blue), loops (white). The same color code for P2 structure in (A) has been used for the corresponding model in (B).
The two hydrophilic portions 66–72 and 95–100 not recognized by proteases were identified as located within two highly structured regions corresponding to the helix α2 and the strand β5, respectively, thus explaining their resistance to protease action. The third protease-resistant region 40–51 is located within a large loop connecting helix α1 and strand β2 and was therefore expected to be proteolytically accessible.
However, the specific amino acid residues occurring in this segment are poorly amenable to proteolysis, causing this portion to be left untouched. It should be noted that the amino acid sequence of this segment shows the lowest degree of similarity with Tomo D.
Finally, unexpected cleavages were observed at Phe101, Leu103, and Trp105 within strand β6, indicating that this region might be endowed with considerable conformational flexibility. It should be noted that the C-terminal portion of P2 does not belong to the regions structurally conserved among the eight clustered NMR structures.
When the data collected from the limited proteolysis experiments carried out on Tomo D were located on the protein model obtained from the Swiss Prot Server (Fig. 3B ▶), an intriguing picture emerged. Most of the preferential cleavage sites identified on P2 found their homologous counterparts in Tomo D (Fig. 1 ▶), whereas few discrepancies could be observed. In particular, the regions Arg25-Asp28 and Asp61-Glu62 occur in exposed loops connecting the end of strand β1 with the first α-helix and strand β4 with the following helix α2, respectively. Accordingly, these portions were found accessible to proteases. The sequences containing these segments are well aligned with P2 protein, and there are no insertions or deletions between the two proteins. Thus, it is surprising that Tomo D is cut by the proteolytic enzymes, whereas P2 is not. Moreover, the flexible loop connecting α1 with strand β2 in Tomo D (region 40–51) contains a number of putative cleavage sites, and yet it was not recognized by proteases.
Given these discrepancies, we also verified whether alternative models of Tomo D structure based on the structures of methane monooxygenase regulatory components, PDB codes 1CKV and 2MOB, would be more consistent with the proteolytic patterns, but this was not the case (data not shown).
Thus, it is evident that there is a discrepancy between the model of the three-dimensional structure of Tomo D and the results of the partial proteolysis experiments. This cannot be due to a difference in flexibility or underdetermination of the NMR structure of P2, since these regions belong to the conserved core. It is then possible that the automatically derived sequence alignment used to build up the model does not correspond to the optimal structural alignment.
Several attempts to obtain an alignment more consistent with the data using different algorithms and scoring systems and including other members of the family did not provide a satisfactory answer. It is obviously possible to manually modify the alignment according to the experimental data. Unfortunately, the constraints given by the proteolytic cleavages do not allow discrimination between a number of alternative alignments. As an example, we show one of them in Figure 4 ▶. This alignment is not optimal in terms of sequence identity (25% with respect to 37% of the automatic alignment) and would imply an insertion immediately after the first β-strand of P2 and a deletion of the irregular region following helix α1. However, the inserted region 25–35 would be exposed to the solvent and would not have any structurally equivalent counterpart in P2, consistent with the proteolysis data.
Fig. 4.

Suboptimal sequence alignment obtained by manual modification of the alignment shown in Fig.1 ▶. The sites of proteolytic cleavages are highlighted in red.
The discrepancy observed at the level of Asp61 and Glu62 might in principle be explained on the basis of the different character of the amino acid residues occurring at homologous positions in P2, that is, Lys and Arg, which can be involved in different interactions. However, neither the structure of P2 nor the Tomo D model supports this hypothesis, as these two residues are exposed and not involved in hydrogen bonds or salt bridges. A more convincing explanation might be given by considering that in P2, the Lys and Arg residues lie quite close to the N-terminal tail, the C-α distance between Lys and residue 4 being about 5 Å. As judged from the NMR data, this part of the protein is flexible and not anchored to the core of the structure, and might hinder the Lys and Arg residues, thus preventing proteolytic cleavages. On the other hand, the long N-terminal extension (16 residues) occurring in Tomo D is most likely structured as being not accessible to proteases (cleavages were only observed at Leu8 and Asp12) and should then not be able to impair cleavages at residues 61 and 62.
Thus, the analysis of the new proposed sequence alignment and the experimental data suggest that the structural similarity between Tomo D and P2 is higher in the C-terminal portion and that the N-terminal region including the end of the first strand, the first α helix and the loop connecting this helix to the second β strand is differently folded in the two proteins.
It is well known that a major problem in predicting protein structure by homology modeling is that the sequence alignment from which the model is built may not be the best with respect to the correct equivalence of residues assessed by structural or functional criteria. It has been proposed (Saqi et al. 1992) that a number of suboptimal alignments should be generated and examined before selecting the appropriate one for model building. In this case, the problem is to find a meaningful procedure to rapidly filter the different possible alignments. This still represents the major obstacle in large-scale automatic modeling, where manual inspection and/or careful analysis of all possible suboptimal sequence alignments is prohibitive.
The results presented in this paper indicate that in this case, standard modeling procedures based on automatic alignment on the structure of P2 fail to produce a model consistent with experimental data. Moreover, this example clearly shows that fast and reliable techniques such as limited proteolysis can be employed to highlight problems in automatic model building, and algorithms should be devised to use this type of experimental data to select among suboptimal alignments.
A further conclusion can be drawn from these data. In all the experiments, a higher enzyme-to-substrate ratio was always needed for P2 with respect to that required for Tomo D, under the same conditions (Table 1). This indicates that, despite the high structural flexibility of P2, Tomo D is even more flexible, and adaptive to its hydroxylase partner. This is in line with the wider substrate specificity of the complex Tomo with respect to other multicomponent monooxygenases (Bertoni et al. 1996). In fact, it has been proposed (Qian et al. 1996) that the flexible cavity defined by helix 2 and strands 3 and 5 is the site where the substrate enters the complex of phenol hydroxylase. If the same functional role is fulfilled by the cavity present in all the models derived for the subunit D of Tomo, then its greater flexibility with respect to that of P2 would allow a different variety of aromatic substrates to entry into the core of the complex.
Materials and methods
Materials
The DNA coding for DmpM inserted into vector pET3a (pET3a-dmpM) was provided by Dr. Justin Powlowski. E. coli strain JM109 was purchased from Boehringer. Labeled oligonucleotides were from Amersham Italia. The Wizard DNA purification kit for elution of DNA fragments from agarose gel was obtained from Promega Italia. Enzymes and other reagents for DNA manipulation were from Promega Italia. PVDF membranes were from Perkin Elmer. Cyanogen bromide, acetic anhydride, trypsin, endoproteinase V-8, chymotrypsin, elastase, and subtilisin were purchased from Sigma. Endoproteinase Asp-N was from Boehringer. Solvents were HPLC-grade from Baker.
General procedures
Bacterial cultures, plasmid purifications, and transformations were performed according to Sambrook et al. (1989). Double-strand DNA was sequenced with the dideoxy method of Sanger et al. (1977), carried out with a Sequenase version II Kit (Amersham Italia) with deoxynucleotide triphosphates purchased from Pharmacia Italia (Italy).
Expression and purification of subunit D of Tomo and DmpM
Aliquots of 20 ng of each plasmid (pMZ1204) (Bertoni et al. 1998) and pET3a-DmpM (Cadieux and Powlowski 1999) were used to transform competent E. coli JM109 cells or BL21(DE3), respectively, plated onto LB/ampicillin plates. One recombinant clone from each transformation was grown in 10 mL of LB-medium, supplemented with ampicillin, at 37°C up to O.D.600 nm = 0.7. These cultures were used to inoculate 1 L of LB supplemented with 50 μg/mL ampicillin, and grown at 37°C until A600 ranged from 0.6 to 0.7. Expression of the recombinant protein was induced by IPTG at a final concentration of 0.5 mM, and the culture was kept for another 3 h at 37 °C under vigorous shaking. Cells were then collected by centrifugation for 10 min at 5,520 × g, at 4°C, and the cell paste stored at −80°C. SDS-PAGE analysis of an aliquot of induced and noninduced cells extracted in electrophoresis loading buffer showed that in each culture, a protein with a molecular mass of about 12 kDa, the expected molecular size of recombinant subunit D and of DmpM, was produced only in the induced cells.
The identity of the expressed proteins was further checked by N-terminal sequence determination, run on samples directly blotted on PVDF membranes from the electrophoretic gel.
Cells from 1-L cultures were suspended in 10 mL of buffer A (25 mM MOPS at pH 6.9, containing 5% glycerol, 1 M DTT) and disrupted by sonication (6 × 1 min cycle, on ice). The soluble fraction was separated by the insoluble fraction by centrifugation at 17,400 × g for 60 min at 4°C. SDS-PAGE analysis of the soluble and insoluble fractions (data not shown) revealed that the proteins of interest were present only in the soluble fraction. Yields were of ∼20–30 mg of protein/L of bacterial culture, on the basis of a densitometric scanning of the electrophoretic profile.
The crude extracts were loaded onto a Fast Flow Q-Sepharose column (10 × 200 mm), equilibrated in buffer A. Elution was carried out at 4°C, at a flow rate of 7 mL/h, with a linear gradient (0 to 0.5 M, 160 mL) of NaCl. The proteins of interest were identified in the eluate by running SDS-PAGE of alternate fractions, and then pooled. Protein samples were concentrated by ultrafiltration in an Amicon apparatus equipped with YM3 membrane, then loaded on a Sephadex G75 superfine column (28 × 350 mm), and eluted at 12 mL/h with buffer A containing 0.3 M NaCl. The analysis of the eluate carried out by SDS-PAGE of alternate fractions revealed that either in the case of DmpM or of subunit D, two distinct peaks, both containing a protein of the expected molecular weight (about 10 kDa in the case of DmpM, and about 12 kDa in the case of subunit D), can be separated (data not shown). It has already been shown (Cadieux and Powlowski 1999) that DmpM exists in two forms: an active monomeric form, and an inactive dimer, which can convert one into the other. Thus, we discarded in both cases the fast-eluting peak and collected the fractions eluting at the correct (monomeric) elution volume.
Samples of purified subunit D and DmpM were desalted by RP-HPLC on a Phenomenex C18 column (see Protein sequence determination section below) and subjected to electrospray mass spectrometry. The average molecular mass values were 12,142.2 ± 0.6 Da, and 10,360.1 ± 0.9 Da, for Tomo D and DmpM, respectively. These values are in agreement with the theoretical values calculated on the basis of the deduced amino acid sequence of subunit D (12,141.8 Da), and that of DmpM (10,359.7 Da), respectively, and confirm that the protein species purified through the procedure above are in their monomeric state. However, given the possibility of the conversion of these species to "oligomeric" forms (Cadieux and Powlowski 1999), we always stored the proteins in glycerol containing buffers, which have been shown (Cadieux and Powlowski 1999) to prevent interconversion of the two forms. Finally, we routinely checked the aggregation state of the samples prior to their use by running analytical gel filtration chromatography on a HiLoad 10/30 Superdex 75 column, eluted with 50 mM ammonium acetate at pH 5.0, containing 0.3 M NaCl, at a flow rate of 0.3 ml/min.
Protein sequence determination
Proteins were desalted on Phenomenex Jupiter C18 reverse-phase columns (250 × 2.1 mm, 100 Å pore size) eluted at 0.2 mL/min with a linear gradient of a two-solvent system. Solvent A was 0.1% TFA in water, solvent B was acetonitrile containing 0.1% TFA. The gradient was constructed increasing the concentration of solvent B from 20% to 60% in 50 min. Desalted proteins (2 nmol) were incubated in 100 μL of a 10-fold molar excess of CNBr dissolved in 70% TFA, for 12 h under nitrogen in the dark. Reaction was stopped by dilution with 1 mL of cold water.
Digestion with endoprotease Asp-N was carried out in 50 mM NaHCO3 at pH 8.0, overnight at 37°C, using a 1 : 50 w : w, enzyme-to-substrate ratio.
Peptide mixtures were directly analyzed by MALDI mass spectrometry.
Limited proteolysis experiments
Limited proteolysis experiments were carried out on 3 nmol of proteins not subjected to the desalting procedure described in the previous paragraph, with trypsin, V-8 protease, endoproteinase Asp-N, chymotrypsin, elastase, and subtilisin. Enzymatic digestions were all performed in 0.4 % ammonium bicarbonate at pH 7.0, at 37°C with enzyme-to-substrate ratios ranging from 1 : 1500 to 1 : 30 w : w (see Table 1). The extent of digestion was monitored on a time-course basis by sampling the reaction mixture at different time intervals from 10 to 60 min. Digested protein samples were acidified by adding TFA to lower the pH to 2.5, and analyzed by RP-HPLC as described in the section on protein sequence determination. Elution was monitored at 220 and 280 nm. Individual fractions were collected and identified by ES/MS.
Mass spectrometry
Protein samples or proteolytic fragments were analyzed by electrospray mass spectrometry using either a BIO-Q triple quadrupole mass spectrometer (Micromass) or an API-100 single quadrupole instrument (Perkin Elmer). Samples were directly injected into the ion source via a loop injection at a flow rate of 5 μl/min. Data were acquired and elaborated using either the MASS-LINX (Micromass) or the Biomultiviewer (Perkin Elmer) program.
Mass calibration was performed by means of the multiply charged ions from a separate injection of horse heart myoglobin (average molecular mass 16,951.5 Da). All masses are reported as average mass. Peptide mixtures were analyzed by MALDI mass spectrometry (MALDI/MS) using a Voyager DE instrument (Perkin Elmer). Typically, 1 μL of analyte solution was mixed with 1 μL of α-cyano-4-hydroxycinammic acid 10 mg/mL in acetonitrile/0.2% TFA, 70 : 30 v/v, containing 250 fmoL of insulin. The mixture was applied onto the metallic sample plate and air dried. Mass calibration was performed with the mass signals of insulin at m/z 5734.5 and a matrix peak at m/z 379.1. Raw data were analyzed by using a computer program provided by the manufacturer. All mass values are reported as average masses.
Sequence alignment and modeling
Protein sequences were aligned using the Fasta3 program (Pearson and Lipman 1988) available on-line at http://www2.ebi.ac.uk/fasta3/.
Protein modeling was carried out with the automated comparative protein server freely available at http://www.expasy.ch/swissmod/SWISS-MODEL.html (Peitsch 1996; Guex and Peitsch 1997). Models were energy-minimized with Gromos96.
Other methods
N-terminal protein sequence determinations were performed on an Applied Biosystems sequenator (model 473A), connected on-line with an HPLC apparatus for identification of phenylthiohydantoins. SDS-PAGE was carried out according to Laemmli (1970). Protein concentrations were determined by a colorimetric assay (BCA Protein Assay, Pierce).
Acknowledgments
The authors are indebted to Dr. Justin Powlowski, Dept. of Chemistry and Biochemistry, Concordia University, Canada for having kindly provided the DNA coding for DmpM, Dr. Antimo Di Maro for the determination of the N-terminal sequence of the proteins, Dr. Alessia Palmieri for contributing to the preparation and characterization of Tomo D, and Mrs. Aida Milano for her contribution in the purification of DmpM. Special thanks go to Prof. Giuseppe D'Alessio, Dept. of Organic and Biological Chemistry, University of Naples Federico II, Italy, for critically reading the manuscript. This work was supported by grants from the Ministry of University and Research (PRIN/98, CSEB:O&O, and PRIN/97, Biologia strutturale).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
Tomo, toluene/o-xylene-monooxygenase
Tomo D, subunit D of the complex Tomo
DmpM (protein P2), regulatory component of phenol hydroxylase
ORF, open reading frame
ES/MS, electrospray mass spectrometry
MALDI, matrix assisted laser desorption ionization
LB, Luria-Bertani
EDTA, ethylenediaminetetraacetic acid
PAGE, polyacrylamide gel electrophoresis
SDS, sodium dodecylsulfate
Tris, tris(hydroxymethyl)aminomethane
DTT, dithiothreitol
HPLC, high pressure liquid chromatography
IPTG, isopropyl-β-D-thiogalacto-pyranoside
PVDF, polyvinyl difluoro
PDB, Protein Data Bank
V-8, V-8 endoproteinase
Asp-N, endoproteinase Asp-N
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.35701.
References
- Bertoni, G., Bolognesi, F., Galli, E., and Barbieri, P. 1996. Cloning of the genes for and characterization of the early stages of toluene catabolism in Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 62 3704–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni, G., Martino, M., Galli, E., and Barbieri, P. 1998. Analysis of the gene cluster encoding toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 64 3626–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne, A.M., Kukor, J.J., and Olsen, R.H. 1995. Sequence analysis of the gene cluster encoding toluene-3-monooxygenase from Pseudomonas Picketii PKO1. Gene 154 65–70. [DOI] [PubMed] [Google Scholar]
- Cadieux, E. and Powlowski, J. 1999. Characterization of active and inactive forms of the phenol hydroxylase stimulatory protein DmpM. Biochemistry 38 10714–10722. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Polverino de Laureto, P., De Filippis, V., Scaramella, E., and Zambonin, M. 1997. Probing the partly folded states of proteins by limited proteolysis. Fold. Des. 2:2 R: 17–26. [DOI] [PubMed] [Google Scholar]
- Froland, W.A., Andersson, K.K., Lee, S.K., Liu, Y., and Lipscomb, J.D. 1992. Methane monooxygenase component B and reductase alter the regioselectivity of the hydroxylase component-catalyzed reactions. A novel role for protein-protein interactions in an oxygenase mechanism. J. Biol. Chem. 267 17588–17597. [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. Swiss-Model and the Swiss-PDB-viewer: An environment for comparative protein modelling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. and Sander, C. 1983. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Laemmli, U. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Moult, J., Hubbard, T., Bryant, S.H., Fidelis, K., and Pedersen, J.T. 1999. Critical assessment of methods of protein structure prediction (CASP): Round II. Proteins: Structure, Function and Genetics S1 2–6. [PubMed] [Google Scholar]
- Newman, L.M. and Wackett, L.P. 1995. Purification and characterization of Toluene 2-monooxygenase from Burkholderia cepacia G4. Biochemistry 34 14066–14076. [DOI] [PubMed] [Google Scholar]
- Nordlund, I., Powlowski, J., and Shingler, V. 1990. Complete nucleotide sequence and polypeptide analysis of multicomponent phenol hydroxylase from Pseudomonas sp. strain CF600. J. Bacteriol. 172 6826–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrù, S., Dal Piaz, F., Casbarra, A., Biasiol, G., De Francesco, R., Steinkühler, C., and Pucci, P. 1999. Conformational changes in the NS3 protease from hepatitis C virus strain Bk monitored by limited proteolysis and mass spectrometry. Protein Sci. 8 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, W.R. and Lipman, D.J. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peitsch, M.C. 1996. Promod and Swiss-Model: Internet-based tools for automated comparative protein modelling. Biochem. Soc. Trans. 24 274–279. [DOI] [PubMed] [Google Scholar]
- Pikus, J.D., Studts, J.M., Achim, C., Kauffmann, K.E., Munck, E., Steffan, R.J., McClay, K., and Fox, B.G. 1996. Recombinant toluene-4-monooxygenase: Catalytic and Mossbauer studies of the purified diiron and rieske components of a four-protein complex. Biochemistry 35 9106–9119. [DOI] [PubMed] [Google Scholar]
- Qian, H., Edlund, U., Powlowski, J., Shingler, V., and Sethson, I. 1996. Solution structure of phenol hydroxylase protein component P2 determined by NMR spectroscopy. Biochemistry 36 495–504. [DOI] [PubMed] [Google Scholar]
- Ryoo, D., Shim, H., Canada, K., Barbieri, P., and Wood, T.K. 2000. Aerobic degradation of tetrachloroethylene by toluene-o-xylene monooxygenase of Pseudomonas stutzeri OX1. Nature Biotechnology 18 775–778. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular Cloning. A Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Sanger, F., Nicklen, S., and Coulson, A.R. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 76 5653–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaloni, A., Miraglia, N., Orrù, S., Amodeo, P., Motta, A., Marino, G., and Pucci, P. 1998. Topology of Calmodulin-Melittin complex. J. Mol. Biol. 277 945–958. [DOI] [PubMed] [Google Scholar]
- Stainthorpe, A.C., Lees, V., Salmond, G.P.C., Dalton, H., and Murrel, J.C. 1990. The methane monoxygenase gene cluster of Metilococcus capsulatus (bath). Gene 91 27–34. [DOI] [PubMed] [Google Scholar]
- Yen, K.-M. and Karl, M.R. 1992. Identification of a new gene, TMOF, in the Pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 174 7253–7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen, K.-M., Karl, M.R., Blatt, M.L., Simon, M.J., Winter, R.B., Fausset, P.R., Lu, H.S., Harcourt, A.A., and Chen, K.K. 1991. Cloning and characterization of a Pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 173 5315–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappacosta, F., Pessi, A., Bianchi, E., Venturini, S., Sollazzo, M., Tramontano, A., Marino, G., and Pucci, P. 1996. Probing the tertiary structure of proteins by limited proteolysis and mass spectrometry. The case of minibody. Protein Sci. 5 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]