

The peripheral membrane flavoprotein pyruvate oxidase from E. coli has been crystallized in the full-length form and as a proteolytically activated truncation variant lacking the last 23 amino acids at the C-terminus.
Keywords: peripheral membrane proteins, thiamine diphosphate, flavin adenine dinucleotide, ubiquinone, pyruvate, electron transfer, decarboxylation
Abstract
The thiamine diphosphate- and flavin-dependent peripheral membrane enzyme pyruvate oxidase from Escherichia coli (EcPOX) has been crystallized in the full-length form and as a proteolytically activated C-terminal truncation variant which lacks the last 23 amino acids (Δ23 EcPOX). Crystals were grown by the hanging-drop vapour-diffusion method using either protamine sulfate (full-length EcPOX) or 2-methyl-2,4-pentanediol (Δ23 EcPOX) as precipitants. Native data sets were collected at a X-ray home source to a resolution of 2.9 Å. The two forms of EcPOX crystallize in different space groups. Whereas full-length EcPOX crystallizes in the tetragonal space group P43212 with two monomers per asymmetric unit, the crystals of Δ23 EcPOX belong to the orthorhombic space group P212121 and contain 12 monomers per asymmetric unit.
1. Introduction
Pyruvate oxidase from Escherichia coli (EcPOX; EC 1.2.2.2) is a peripheral membrane enzyme that consists of four identical subunits of M r 62 000, each of which contains one tightly bound flavin adenine dinucleotide (FAD), one thiamine diphosphate (ThDP) and a divalent metal ion (Mg2+) per active site (Williams & Hager, 1966 ▶; O’Brien et al., 1977 ▶; Blake et al., 1982 ▶). EcPOX catalyzes the oxidative decarboxylation of pyruvate to acetate and carbon dioxide (Hager, 1957 ▶). The reducing equivalents which arise during the oxidation of pyruvate at the thiamine site are initially transferred to the neighbouring flavin cofactor. In the reduced state, the enzyme adheres to the biological membrane in E. coli and eventually transfers both electrons to ubiquinone-8 (Q8; Cunningham & Hager, 1975 ▶; Marchal et al., 2001 ▶), a membrane-bound mobile carrier of the electron-transport chain. The enzyme exhibits a very low basal activity that can be stimulated several hundred-fold either by binding to lipid amphiphiles or alternatively, under in vitro conditions, by mild limited proteolysis (Cunningham & Hager, 1971a ▶,b ▶; Russell et al., 1977 ▶). Treatment of full-length EcPOX with α-chymotrypsin results in the cleavage of a 23-residue peptide (α-peptide) from the C-terminus of each monomer. Both activation methods (membrane/lipid binding and proteolysis) yield fully active enzyme with similar kinetic properties.
As the α-peptide has been shown to bind to phospholipid vesicles, it has been concluded that the C-terminal residues of EcPOX mediate membrane binding (Zhang & Hager, 1987 ▶).
Here, we describe the crystallization and preliminary X-ray diffraction analysis of the non-activated full-length protein (residues 1–572) and the proteolytically activated Δ23 variant (residues 1–549) as part of a study towards understanding the molecular details of the membrane-binding mechanism and concomitant enzyme activation of EcPOX.
2. Materials and methods
2.1. Protein purification
Pyruvate oxidase from E. coli was overexpressed in E. coli strain ZK126 carrying the plasmid pYYC102 which encodes the poxB gene (the plasmid and strain were kindly provided by John E. Cronan Jr, University of Illinois; Wang et al., 1991 ▶). The cells were grown in LB medium supplemented with 0.1 mg ml−1 ampicillin at 310 K for 19 h. Expression of poxB was induced at the early stationary phase by the rpoS gene-encoded σS factor. Cells were harvested with a Beckman centrifuge (J2-HC, JA-10 rotor; Beckman Coulter Inc., USA) at 4000g for 10 min at 277 K, flash-frozen and stored at 253 K until use.
For purification, about 20 g of cells was thawed on ice and resuspended in two volumes of 20 mM potassium phosphate buffer pH 6.5 supplemented with 0.1 mM FAD. The cells were then disrupted by repeated passage through a French Press apparatus (Gaulin, APV Homogeniser GmbH, Germany) at 120 MPa. Cell debris was separated from the soluble fraction by ultracentrifugation at 70 000g for 30 min at 277 K in a Beckman L8-M ultracentrifuge using a 45-Ti rotor. Nucleic acids were precipitated with 0.5%(w/v) streptomycin sulfate for 30 min at 281 K. After subsequent ultracentrifugation (using the same conditions as above), the clear supernatant was heated for 2 min at 338 K in a water bath. Denatured thermolabile proteins were separated by ultracentrifugation (using the same conditions as above) and the remaining bright yellow supernatant was loaded onto an anion-exchange column (HiLoad Q Sepharose 26/60, GE Healthcare, Sweden) previously equilibrated with 20 mM potassium phosphate buffer pH 6.5. For elution, a linear gradient of 20–300 mM potassium phosphate buffer pH 6.5 over six column volumes at a flow rate of 1.5 ml min−1 was employed. Pyruvate oxidase eluted at approximately 160 mM potassium phosphate. Fractions that contained a sufficient amount of enriched EcPOX were pooled and concentrated by ultrafiltration (Amicon Ultra-15, Millipore, USA) to a total volume of approximately 4 ml. The concentrated protein was then applied onto a gel-filtration column (Superdex 200 26/60, GE Healthcare, Sweden) previously equilibrated with 300 mM potassium phosphate buffer pH 6.5 and eluted in the tetrameric form using the same buffer at a flow rate of 0.9 ml min−1. The purity of the enzyme was analyzed by SDS–PAGE according to the method of Laemmli (1970 ▶). The fractions with the highest homogeneity (>95%) were pooled and the buffer was exchanged to 20 mM potassium phosphate pH 6.0 by ultrafiltration (Amicon Ultra-15, Millipore, USA). The total amount of homogeneous protein varied between 20 and 25 mg per litre of cultivation medium.
The protein concentration was estimated according to the method of Bradford (1976 ▶). The enzymatic activity of the purified full-length EcPOX was determined in the absence and presence of lipid amphiphiles using ferricyanide (∊450 = 218.8 M −1 cm−1) as an artificial electron acceptor (Mather & Gennis, 1985 ▶). The protein fractions with the highest specific activity were either directly used for crystallization setup and limited proteolysis or flash-frozen in liquid nitrogen and stored at 253 K.
2.2. Limited proteolytic digestion
For in vitro activation of purified full-length EcPOX, the protein was subjected to limited proteolysis using α-chymotrypsin, which cleaves off the C-terminal 23-residue α-peptide from each monomer. The proteolytic digestion was carried out at room temperature (298 K) according to an established protocol detailed in Recny & Hager (1983 ▶) with some slight modifications. A typical reaction mixture contained 3 mg ml−1 EcPOX reconstituted with 20 mM MgSO4 and 10 mM ThDP in 100 mM potassium phosphate buffer pH 6.0. After an incubation time of 5 min, EcPOX was reacted with 200 mM pyruvate for 10 min, resulting in complete reduction of the enzyme-bound flavin. The proteolytic digestion was initiated by addition of 20 µg ml−1 α-chymotrypsin (1 mg ml−1 stock solution in 1 mM HCl). The proteolysis reaction was stopped after 40 min incubation time by the addition of a tenfold molar excess of aprotinin, a protease inhibitor, with respect to the protease concentration employed. The reaction mixture was immediately ultrafiltrated (Amicon Ultra-4, Millipore, USA) for 5 min at 2600g at 281 K to quantitatively remove chymotrypsin and the α-peptide from the C-terminally truncated EcPOX. After this short ultrafiltration step, the protein was resuspended in 20 mM potassium phosphate buffer pH 6.0 supplemented with 20 mM MgSO4, 5 mM ThDP and 0.1 mM FAD and subjected to repeated cycles of ultrafiltration. Excess cofactors were then separated by diafiltration using 20 mM potassium phosphate pH 6.0. The enzymatic activity of the proteolytically processed EcPOX was determined in an artificial redox assay as detailed above for the full-length enzyme.
3. Results and discussion
3.1. Crystallization
Full-length EcPOX and the C-terminal Δ23 truncation variant were successfully crystallized by the hanging-drop vapour-diffusion method (McPherson, 1982 ▶). In initial crystallization trials with full-length EcPOX, we tested the conditions reported by Williams and Hager, who observed spontaneous crystallization of EcPOX during purification (Williams & Hager, 1966 ▶). Further crystallization screenings revealed a reservoir mixture of 80 mM potassium phosphate buffer pH 6.0 supplemented with 0–1%(w/v) polyethylene glycol 2000 and 0.05–0.10%(w/v) protamine sulfate to be optimal for reproducible crystallization. In a typical crystallization setup, 2 µl protein solution (10 mg ml−1 EcPOX in 20 mM potassium phosphate pH 6.0 plus 10 mM ThDP and 10 mM MgSO4) was mixed with 2 µl of the appropriate reservoir solution. Drops were equilibrated against 500 µl reservoir volume. Yellow crystals of full-length EcPOX (Fig. 1 ▶) grew within two weeks at 281 K.
Figure 1.

Representative crystal of full-length EcPOX.
Initial crystallization screenings with the C-terminally truncated Δ23 EcPOX were carried out in a 96-well sitting-drop plate. Several different conditions from these screens gave small yellow crystals. Further optimization of these conditions in a 24-well hanging-drop plate revealed 100 mM MES–NaOH pH 6.2 and 20–35% 2-methyl-2,4-pentanediol (MPD) to be most effective for crystallization. Typically, 2 µl of this reservoir solution was mixed with 2 µl protein solution (10 mg ml−1 Δ23 EcPOX in 20 mM potassium phosphate pH 6.0 with 10 mM ThDP and 10 mM MgSO4) and equilibrated against a 500 µl reservoir volume. Bright yellow crystals of Δ23 EcPOX (Fig. 2 ▶) grew within two weeks at 286 K.
Figure 2.

Representative crystal of Δ23 EcPOX.
3.2. Data collection and preliminary X-ray diffraction analysis
Crystals of full-length EcPOX were mounted in cryoloops (Hampton Research, USA) and subjected to a 10 s soak in 80 mM potassium phosphate buffer pH 6.0 supplemented with 0.1% protamine sulfate and 17% ethylene glycol. Subsequently, the crystals were soaked again for 10 s in a similar solution but containing 29% ethylene glycol, immediately flash-cooled by direct immersion into liquid nitrogen and transferred to the goniometer head. The crystals of the Δ23 EcPOX needed no further cryoprotection as they were grown in 30% MPD.
Diffraction data for both full-length and Δ23 EcPOX were collected in-house in a 100 K nitrogen cryostream (XSTREAM2000, Rigaku/MSC, Japan) with an R-AXIS IV++ imaging-plate system (Rigaku/MSC, Japan) using Cu Kα radiation (wavelength 1.5418 Å) generated by a Rigaku MM-007 rotating-anode generator. The data sets were initially processed and scaled with XDS (Kabsch, 1993 ▶). Data-collection statistics are summarized in Table 1 ▶. Full-length EcPOX crystallized in the tetragonal space group P43212 with two monomers per asymmetric unit, whereas crystals of the Δ23 truncation variant belong to the orthorhombic space group P212121 and contain 12 monomers (three tetramers) per asymmetric unit. Preliminary molecular-replacement phasing (MR) of full-length EcPOX and Δ23 EcPOX was carried out with Phaser (McCoy et al., 2005 ▶) using the structure of the related pyruvate oxidase from Lactobacillus plantarum (LpPOX; PDB code 1pow) as a search model. Full-length EcPOX and LpPOX share 29% sequence identity and 47% sequence similarity and can thus be anticipated to possess similar three-dimensional structures. Structural analysis of EcPOX promises to provide detailed insights into the mechanism of membrane binding as well as of enzymatic catalysis, including reaction intermediates (Wille et al., 2006 ▶; Tittmann et al., 2005 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Full-length EcPOX | Δ23 EcPOX | |
|---|---|---|
| Space group | P43212 | P212121 |
| Molecules per ASU | 2 | 12 |
| Unit-cell parameters | ||
| a (Å) | 151.18 | 198.04 |
| b (Å) | 151.18 | 201.95 |
| c (Å) | 150.71 | 209.84 |
| Mosaicity (°) | 0.19 | 0.22 |
| Resolution range (Å) | 29.09–2.9 | 19.89–2.9 |
| Total no. of reflections | 235811 | 461798 |
| Unique reflections | 37553 | 162034 |
| Redundancy | 6.28 | 2.85 |
| Completeness (%) | 95.4 (96.8) | 96.4 (96.5) |
| 〈I/σ(I)〉 | 14.54 (3.43) | 7.21 (2.49) |
| Rmerge† (%) | 11.3 (54.5) | 14.5 (45.8) |
| Wilson B factor (Å2) | 58.8 | 46.2 |
R
merge = 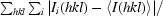
 .
.
Acknowledgments
This work was supported by the DFG Graduiertenkolleg 1026 ‘Conformational Transitions in Macromolecular Interactions’. We thank John Cronan Jr for providing plasmid pYYC102.
References
- Blake, R., O’Brien, T., Gennis, R. & Hager, L. (1982). J. Biol. Chem.257, 9605–9611. [PubMed]
- Bradford, M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed]
- Cunningham, C. & Hager, L. (1971a). J. Biol. Chem.246, 1575–1582. [PubMed]
- Cunningham, C. & Hager, L. (1971b). J. Biol. Chem.246, 1583–1589. [PubMed]
- Cunningham, C. & Hager, L. (1975). J. Biol. Chem.250, 7139–7146. [PubMed]
- Hager, L. (1957). J. Biol. Chem.229, 251–263. [PubMed]
- Kabsch, W. (1993). J. Appl. Cryst.26, 795–800.
- Laemmli, U. K. (1970). Nature (London), 227, 680–685. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- McPherson, A. (1982). Preparation and Analysis of Protein Crystals. New York: Wiley.
- Marchal, D., Pantigny, J., Laval, J., Moiroux, J. & Bourdillon, C. (2001). Biochemistry, 40, 1248–1256. [DOI] [PubMed]
- Mather, M. & Gennis, R. (1985). J. Biol. Chem.260, 10395–10397. [PubMed]
- O’Brien, T. A., Blake, R. & Gennis, R. B. (1977). Biochemistry, 16, 3105–3109. [DOI] [PubMed]
- Recny, M. A. & Hager, L. (1983). J. Biol. Chem.258, 5189–5195. [PubMed]
- Russell, P., Hager, L. & Gennis, R. B. (1977). J. Biol. Chem.252, 7877–7882. [PubMed]
- Tittmann, K., Wille, G., Golbik, R., Weidner, A., Ghisla, S. & Hübner, G. (2005). Biochemistry, 44, 13291–13303. [DOI] [PubMed]
- Wang, A., Chang, Y. & Cronan, J. Jr (1991). J. Biol. Chem.266, 10959–10966. [PubMed]
- Wille, G., Meyer, D., Steinmetz, A., Hinze, E., Golbik, R. & Tittmann, K. (2006). Nature Chem. Biol.2, 324–328. [DOI] [PubMed]
- Williams, F. & Hager, L. (1966). Arch. Biochem. Biophys.116, 168–176. [DOI] [PubMed]
- Zhang, T. & Hager, L. (1987). Arch. Biochem. Biophys.255, 485–487. [DOI] [PubMed]