

Polyketide synthase-1 from C. sativa has been crystallized. The crystal diffracted to 1.55 Å resolution with sufficient quality for further structure determination.
Keywords: cannabinoids, polyketide synthase, hexanoyl triacetic acid lactone
Abstract
Polyketide synthase-1 (PKS-1) is a novel type III polyketide synthase that catalyzes the biosynthesis of hexanoyl triacetic acid lactone in Cannabis sativa (Mexican strain). PKS-1 was overproduced in Escherichia coli, purified and finally crystallized in two different space groups. The crystal obtained in 0.1 M HEPES buffer pH 7.5 containing 0.2 M calcium acetate and 20%(w/v) polyethylene glycol 3350 diffracted to 1.65 Å resolution and belonged to space group P1, with unit-cell parameters a = 54.3, b = 59.3, c = 62.6 Å, α = 69, β = 81, γ = 80°. Another crystal obtained in 0.1 M HEPES buffer pH 7.5 containing 0.2 M sodium chloride and 20%(w/v) polyethylene glycol 3350 diffracted to 1.55 Å resolution and belonged to space group P212121, with unit-cell parameters a = 54.3, b = 110, c = 130 Å. These data will enable us to determine the crystal structure of PKS-1.
1. Introduction
Cannabinoids, which are predominantly produced in Cannabis sativa, are unique secondary metabolites consisting of alkylresorcinol and monoterpene groups. More than 60 cannabinoids have been isolated from marijuana or fresh cannabis leaves and their biosynthetic pathways have been extensively investigated (Turner et al., 1980 ▶; Morimoto et al., 1999 ▶; Shoyama et al., 2005 ▶; Taura, Sirikantaramas, Shoyama, Shoyama et al., 2007 ▶). These studies demonstrated that the major cannabinoids tetrahydrocannabinolic acid and cannabidiolic acid are biosynthesized from the common precursor cannabigerolic acid by novel FAD-dependent oxidases named tetrahydrocannabinolic acid synthase and cannabidiolic acid synthase, respectively (Sirikantaramas et al., 2004 ▶; Taura, Sirikantaramas, Shoyama, Yoshikai et al., 2007 ▶). In addition, Fellermeier and Zenk have identified a geranyltransferase activity producing cannabigerolic acid from olivetolic acid (OLA) and geranylpyrophosphate, demonstrating that the alkylresorcinol moieties of cannabinoids are derived from OLA (Fellermeier & Zenk, 1998 ▶). However, the enzyme catalyzing OLA biosynthesis has neither been identified nor cloned.
OLA, the precursor of cannabinoids, is thought to be one of the polyketides produced via the acetate pathway (Fellermeier et al., 2001 ▶), suggesting that OLA production is catalyzed by polyketide synthase (PKS). Plant PKSs are considered to participate in the biosynthesis of a wide range of biologically important natural products, including OLA (Austin & Noel, 2003 ▶). Despite the diversity of polyketide metabolites, most plant PKSs have high similarity (over 50% sequence identity) to each other. Recent studies based on X-ray crystallographic analysis have demonstrated that various plant PKSs are derived from chalcone synthase, a ubiquitous enzyme in the plant kingdom, by changes in the active-site residues regulating substrate specificity and/or cyclization reactions of linear polyketide intermediates (Ferrer et al., 1999 ▶; Jez et al., 2000 ▶; Austin et al., 2004 ▶; Morita et al., 2007 ▶). In contrast, the mechanism producing polyketides containing phenol carboxylate skeletons such as stilbene carboxylate and OLA is still unclear.
We have recently cloned the gene (GenBank accession No. AB164375) coding for a plant type III polyketide synthase named polyketide synthase-1 (PKS-1) from C. sativa. The amino-acid sequence of PKS-1 showed ∼65% identity to the structurally determined chalcone synthase from Medicago sativa (Ferrer et al., 1999 ▶); however, it contained several amino-acid substitutions in the active site, implying that PKS-1 is a novel enzyme that has evolved from chalcone synthase to catalyze OLA biosynthesis. Our enzymatic assay showed that the PKS-1 did not directly synthesize OLA but produced hexanoyl triacetic acid lactone (HTAL; Fig. 1 ▶), a structural isomer of OLA, from malonyl-CoA and hexanoyl-CoA. Interestingly, a similar enzymatic reaction is catalyzed by a Hydrangea PKS: this PKS, called p-coumaloyl triacetic acid lactone (CTAL) synthase, produces CTAL from malonyl-CoA and p-coumaloyl-CoA (Akiyama et al., 1999 ▶). Austin and coworkers proposed that CTAL is the biosynthetic precursor of stilbene carboxylate (Austin et al., 2004 ▶). Therefore, we also assumed that HTAL is the precursor of OLA and that PKS-1 is involved in OLA biosynthesis (Fig. 1 ▶). To investigate the structure–function relationships of PKS-1 at the atomic level, we have attempted structural characterization of this enzyme by X-ray crystallography. Here, we describe the crystallization and preliminary X-ray diffraction studies of PKS-1.
Figure 1.

PKS-1-catalyzed biosynthesis of hexanoyl triacetic acid lactone (HTAL).
2. Methods
2.1. Expression and purification
Recombinant PKS-1 was expressed in Escherichia coli as a soluble protein with a hexahistidine tag at the C-terminus. The coding region for the full-length PKS-1 (residues 1–385) was amplified using a PKS-1 cDNA clone as a template with the primers 5′-CGGGATCCCATATGAATCATCTTCGTGCTG-3′ and 5′-TATCTTGTCGACATATTTGATGGGAACACT-3′. The resulting fragment was cut with the restriction enzymes NdeI and SalI and cloned into pET24a(+) (Invitrogen) to construct an expression vector which directs the expression of PKS-1-Val-Glu-His6. The vector was then transformed into E. coli strain BL21 (DE3) (Invitrogen). Transformed E. coli was grown at 310 K in 500 ml Luria–Bertani (LB) medium containing 25 µg ml−1 kanamycin until the optical density (660 nm) reached 0.6. After induction with 0.4 mM isopropyl β-d-1-thiogalactopyranoside, growth was allowed to continue for an additional 5 h at 298 K. The cells were harvested by centrifugation for 20 min at 4500 rev min−1 and the pellet was resuspended in 20 ml lysis buffer consisting of 20 mM Tris–HCl pH 8.0 containing 500 mM NaCl, 10 mM imidazole and 2 mM dithiothreitol (DTT). The resuspended solution was then sonicated and the supernatant was collected by centrifugation for 15 min at 14 000 rev min−1 and 277 K.
The PKS-1 was directly applied onto an Ni–NTA column (2.0 × 1.5 cm; GE Healthcare) equilibrated with 20 mM Tris–HCl pH 8.0 containing 10 mM imidazole, 500 mM sodium chloride and 2 mM DTT. After washing the column with 20 mM Tris–HCl pH 8.0 containing 50 mM imidazole, 500 mM sodium chloride and 2 mM DTT, the PKS-1 was eluted with 20 mM Tris–HCl pH 8.0 containing 240 mM imidazole, 500 mM sodium chloride and 2 mM DTT. The eluted fraction containing PKS-1 was desalted by gel filtration using PD10 (10 × 50 mm; GE Healthcare) equilibrated with 20 mM Tris–HCl buffer containing 2 mM DTT. The desalted solution containing PKS-1 was applied onto a Resource Q anion-exchange column (1.0 ml; GE Healthcare). After the column had been washed with 5 ml 20 mM Tris–HCl buffer containing 2 mM DTT, the PKS-1 was eluted with a linear gradient from 20 mM Tris–HCl buffer pH 8.0 containing 2 mM DTT to the same buffer containing 500 mM sodium chloride with a flow rate of 1.0 ml min−1 for 30 min. The purity of the recombinant PKS-1 was verified by SDS–PAGE analysis. The molecular mass of the enzyme was determined by MALDI–TOF MS analysis as described previously (Shoyama et al., 1993 ▶).
2.2. Crystallization
Initial screening for PKS-1 crystallization was performed by the sitting-drop vapour-diffusion method at 293 K using 96-well Intelliplates (Hampton Research) and a Hydra II Plus One (Matrix Technology). The protein concentration was 10.0 mg ml−1 in 20 mM Tris–HCl pH 8.0 containing 150 mM sodium chloride and 2 mM DTT. A sitting drop was prepared by mixing 0.3 µl each of the protein solution and the reservoir solution; the resulting drop was equilibrated against 95 µl reservoir solution. The initial search for crystallization conditions was performed using the commercially available kits Crystal Screen I and II (Hampton Research) and Wizard Screen I and II (Emerald Biostructures).
Of the 400 conditions screened, several microcrystals were obtained after 2 d from drops containing precipitate using the following four conditions. Cubic shaped crystals were obtained from Crystal Screen II condition No. 38 [20%(w/v) PEG 10 000, 100 mM HEPES pH 7.5]. Thin and layered clusters were obtained from three other conditions: Crystal Screen II condition No. 26 [30%(w/v) polyethylene glycol (PEG) monomethyl ether 5000, 100 mM MES monohydrate pH 6.5, 200 mM ammonium sulfate], Wizard Screen I condition No. 28 [20%(w/v) PEG 3000, 100 mM HEPES pH 7.5, 200 mM sodium chloride] and Wizard Screen II condition No. 18 [20%(w/v) PEG 3000, 100 mM Tris pH 7.0, 200 mM calcium acetate]. Based on these results, crystallization conditions were further optimized by changing the precipitant (from PEG 3000 to PEG 3350), pH value and additives. In addition, the protein solutions were kept under reducing condition using DTT and handling under nitrogen gas for optimal growth of crystals. Finally, crystals suitable for X-ray diffraction studies were obtained under two conditions derived from Wizard Screen I condition No. 18 and Wizard Screen II condition No. 28.
2.3. Data collection, processing and molecular replacement
Data-collection experiments using PKS-1 crystals from the two conditions were performed on beamline 5A at the Photon Factory (Tsukuba, Japan) and on beamline 41XU at SPring-8 (Harima, Japan). Reflections were collected on an Area Detector Systems Corporation Quantum 315 CCD at 100 K using crystals that had been soaked in reservoir solution containing 15%(v/v) glycerol as a cryoprotectant and then flash-frozen in a nitrogen-gas stream. During data collection, the crystals were rotated to cover 180° with 1.0° oscillation per frame. Intensity data were processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). The molecular-replacement analyses were performed using the program AMoRe (Navaza, 1994 ▶) using the coordinates of chalcone synthase from alfalfa (Ferrer et al., 1999 ▶; PDB code 1cgz) as an initial search model without any modifications at 15–3 Å resolution.
3. Results and discussion
Recombinant PKS-1 with a C-terminal hexahistidine tag was expressed in E. coli BL21 (DE3) as a soluble protein and was purified by Ni–NTA and Resource Q column chromatography. The molecular weight of the enzyme measured by MALDI–TOF MS analysis was 43 601 Da, which was in a good agreement with the theoretical value of 43 636 Da calculated for the recombinant form of PKS-1 (PKS-1-Val-Glu-His6), indicating that PKS-1 had been correctly expressed and purified. The final yield of the protein was about 4.8 mg per litre of culture. Following optimization of the crystallization conditions, diffraction-quality crystals were finally obtained under two conditions, 100 mM HEPES buffer pH 7.5 containing 200 mM calcium acetate and 20%(w/v) PEG 3350 (crystal A) and 100 mM HEPES buffer pH 7.5 containing 200 mM sodium chloride and 20%(w/v) PEG 3350 (crystal B), after 2 d incubation at 293 K. The typical crystal sizes were approximately 0.02 × 0.02 × 0.3 mm for crystal A and 0.1 × 0.1 × 0.5 mm for crystal B (Fig. 2 ▶).
Figure 2.

Crystals of PKS-1. (a) Crystal A from 100 mM HEPES buffer pH 7.5 containing 200 mM calcium acetate and 20%(w/v) PEG 3350. (b) Crystal B from 100 mM HEPES buffer pH 7.5 containing 200 mM sodium chloride and 20%(w/v) PEG 3350.
Crystals A and B belonged to space groups P1 and P212121, with unit-cell parameters a = 54.3, b = 59.3, c = 62.6 Å, α = 69, β = 81, γ = 80° and a = 53.2, b = 110, c = 130 Å, respectively. The calculated Matthews coefficients of crystals A and B were approximately 2.2 and 2.3 Å3 Da−1, respectively, assuming the presence of two molecules of PKS-1 in the asymmetric unit. The diffraction data sets from crystals A and B were integrated and scaled to maximum resolutions of 1.65 and 1.55 Å resolution, respectively. Data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection and reduction statistics of PKS-1 crystals.
Values in parentheses are for the highest resolution shell.
| Crystal A | Crystal B | |
|---|---|---|
| Crystal data | ||
| Space group | P1 | P212121 |
| Unit-cell parameters (Å, °) | a = 54.3, b = 59.3, c = 62.6, α = 69, β = 81, γ = 80 | a = 53.2, b = 110, c = 130 |
| Matthews coefficient (Å3 Da−1) | 2.2 | 2.3 |
| No. of molecules in the ASU | 2 | 2 |
| Data collection | ||
| Wavelength (Å) | 1.0000 | 1.0000 |
| Beamline | BL5A, Photon Factory | BL41XU, SPring-8 |
| Resolution (Å) | 58–1.65 (1.71–1.65) | 84–1.55 (1.61–1.55) |
| No. of observed reflections | 148748 | 618630 |
| No. of unique reflections | 77414 | 108156 |
| Redundancy | 1.9 (1.9) | 5.7 (4.0) |
| 〈I/σ(I)〉 | 29.4 (6.5) | 29.3(2.2) |
| Rmerge† | 0.052 (0.137) | 0.059 (0.303) |
| Completeness (%) | 90.4 (90.4) | 97.3 (86.7) |
| Wilson plot B factor (Å2) | 19.4 | 17.7 |
R
merge = 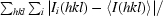
 .
.
Since the amino-acid sequence identity of PKS-1 to that of chalcone synthase from M. sativa (alfalfa) is 65%, the tertiary structure of PKS-1 should be similar to that of chalcone synthase. The locations of two PKS-1 molecules in crystals A and B were identified by molecular-replacement analysis with a correlation F of 0.812 and 0.800 and a crystallographic R factor of 27.7% and 30.0%, respectively. These values were conspicuously better than those for other solutions. In addition to pursuing refinement of the PKS-1 crystal structures, we are preparing to produce complex crystals with substrates or product.
Acknowledgments
We thank Drs Y. Yamada and S. Wakatsuki at Photon Factory (proposal No. 2007G041) and N. Shimizu and M. Kawamoto at SPring-8 (proposal No. 2007B1900) for data collection.
References
- Akiyama, T., Shibuya, M., Liu, H. M. & Ebizuka, Y. (1999). Eur. J. Biochem.263, 834–839. [DOI] [PubMed]
- Austin, M. B., Bowman, M. E., Ferrer, J.-L., Schröder, J. & Noel, J. P. (2004). Chem. Biol.11, 1179–1194. [DOI] [PubMed]
- Austin, M. B. & Noel, J. P. (2003). Nat. Prod. Rep.20, 79–110. [DOI] [PubMed]
- Fellermeier, M., Eisenreich, W., Bacher, A. & Zenk, M. H. (2001). Eur. J. Biochem.268, 1596–1604. [DOI] [PubMed]
- Fellermeier, M. & Zenk, M. H. (1998). FEBS Lett.427, 283–285. [DOI] [PubMed]
- Ferrer, J.-L., Jez, J. M., Bowman, M. E., Dixon, R. A. & Noel, J. P. (1999). Nature Struct. Biol.6, 775–784. [DOI] [PubMed]
- Jez, J. M., Austin, M. B., Ferrer, J.-L., Bowman, M. E., Schröder, J. & Noel, J. P. (2000). Chem. Biol.7, 919–930. [DOI] [PubMed]
- Morimoto, S., Taura, F. & Shoyama, Y. (1999). Curr. Top. Phytochem.2, 103–113.
- Morita, H., Kondo, S., Oguro, S., Noguchi, H., Sugio, S., Abe, I. & Kohno, T. (2007). Chem. Biol.14, 359–369. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Shoyama, Y., Sakata, R., Isobe, R. & Murakami, H. (1993). Org. Mass Spectr.28, 987–988.
- Shoyama, Y., Takeuchi, A., Taura, F., Tamada, T., Adachi, M., Kuroki, R., Shoyama, Y. & Morimoto, S. (2005). Acta Cryst. F61, 799–801. [DOI] [PMC free article] [PubMed]
- Sirikantaramas, S., Morimoto, S., Shoyama, Y., Ishikawa, Y., Wada, Y., Shoyama, Y. & Taura, F. (2004). J. Biol. Chem.279, 39767–39774. [DOI] [PubMed]
- Taura, F., Sirikantaramas, S., Shoyama, Y., Shoyama, Y. & Morimoto, S. (2007). Chem. Biodivers.4, 1649–1663. [DOI] [PubMed]
- Taura, F., Sirikantaramas, S., Shoyama, Y., Yoshikai, K., Shoyama, Y. & Morimoto, S. (2007). FEBS Lett.581, 2929–2934. [DOI] [PubMed]
- Turner, C. E., Elsohly, M. A. & Boeren, E. G. (1980). J. Nat. Prod.43, 169–234. [DOI] [PubMed]