

The complex between the Ets domain of Ets2 and its target DNA has been crystallized. The crystals diffracted to 3.0 Å resolution.
Keywords: transcription factor, Ets domain, protein–DNA complex crystal, NBSD-195
Abstract
The Ets2 transcription factor is a member of the Ets transcription-factor family. Ets2 plays a role in the malignancy of cancer and in Down’s syndrome by regulating the transcription of various genes. The DNA-binding domain of Ets2 (Ets domain; ETSD), which contains residues that are highly conserved among Ets transcription-factor family members, was expressed as a GST-fusion protein. The aggregation of ETSD produced after thrombin cleavage could be prevented by treatment with NDSB-195 (nondetergent sulfobetaine 195). ETSD was crystallized in complex with DNA containing the Ets2 target sequence (GGAA) by the hanging-drop vapour-diffusion method. The best crystals were grown using 25% PEG 3350, 80 mM magnesium acetate, 50 mM sodium cacodylate pH 5.0/5.5 as the reservoir at 293 K. The crystals belonged to space group C2, with unit-cell parameters a = 85.89, b = 95.52, c = 71.89 Å, β = 101.7° and a V M value of 3.56 Å3 Da−1. Diffraction data were collected to a resolution of 3.0 Å.
1. Introduction
The Ets (E26 transformation-specific) transcription-factor family is involved in cellular differentiation, proliferation, senescence, apoptosis and oncogenic transformation by regulating the transcription of various genes (Oikawa & Yamada, 2003 ▶; Sementchenko & Watson, 2000 ▶; Sharrocks, 2001 ▶). Approximately 30 members of the family have been identified in mammals and they contain a highly conserved 85-amino-acid DNA-binding domain termed the Ets domain. They recognize and bind to the consensus GGA core sequence (Ets-binding site) and additional flanking sequences on gene promoters, which determine the binding specificity of each factor (Hollenhorst et al., 2007 ▶). Ets transcription-factor family members can be classified into subfamilies (Ets, TEL, ELF etc.) based on the similarity of the Ets-domain sequences and the domain compositions. Most Ets transcription factors are functional as monomers, unlike many other transcription factors which function as homodimers or heterodimers (Garvie & Wolberger, 2001 ▶). Moreover, Ets-family members regulate gene transcription cooperatively with other transcription factors and cofactors through physical interactions on the promoters (Li et al., 2000 ▶).
Ets2 is a member of the Ets subfamily of the Ets transcription-factor family; this subfamily consists of Ets1 and Ets2. Ets2 is involved in the malignancy of cancer by regulating various genes related to the cell cycle and apoptosis (Dwyer et al., 2007 ▶; Hsu et al., 2006 ▶; Li et al., 2007 ▶; Tynan et al., 2005 ▶). Ets2 has also been implicated in acute myelogenous leukaemia (AML; Baldus et al., 2004 ▶; Sacchi et al., 1988 ▶). When the Ets2 gene located on the distal part of chromosome 21 (21q22.1–22.3) translocates to chromosome 8, the translocation products cause AML (Watson et al., 1985 ▶). Furthermore, Ets2 is overexpressed in Down’s syndrome patients and its activation of p53 and caspase-3 gene transcriptions may result in neuronal apoptosis and skeletal muscle abnormalities (Raouf & Seth, 2000 ▶; Wolvetang, Bradfield et al., 2003 ▶; Wolvetang, Wilson et al., 2003 ▶). Ets2 is phosphorylated at Thr72 by ERK2 in the Ras/MAP kinase cascade and the phosphorylation increases protein stability and transcriptional up-regulation activity (Foulds et al., 2004 ▶; Yang et al., 1996 ▶).
To date, the crystal structures of several Ets domain–DNA complexes have been reported (Escalante et al., 2002 ▶; Garvie et al., 2001 ▶, 2002 ▶; Kodandapani et al., 1996 ▶; Mo et al., 1998 ▶, 2000 ▶; Obika et al., 2003 ▶; Pio et al., 1996 ▶; Wang et al., 2005 ▶). The Ets domain adopts a winged helix–turn–helix (wHTH) protein fold containing three α-helices and four β-strands. The second helix (α3) in the wHTH motif mainly recognizes DNA bases and also interacts with phosphate backbones, as do the N-terminal loop and C-terminal winged moiety of α3. Furthermore, the complex structures reveal the structural basis of the DNA-binding specificity of some Ets-family members. For example, although the Ets domains of Elk1 and Sap1 have highly conserved residues in α3, nearly a third of the interactions between α3 and DNA differ between the Elk1–DNA and Sap1–DNA complexes, resulting in a difference in their DNA-binding ability. The different DNA-binding properties of these proteins are mediated by nonconserved residues distal to the DNA-binding surface that cause a change in the side-chain conformation between Tyr66Elk1 and the corresponding Tyr65Sap1 in α3 (Mo et al., 2000 ▶). However, in the Ets domains of many other Ets-family members the relationships between amino-acid sequence and structure and between amino-acid sequence and DNA-binding ability remain unclear.
The Ets domain of Ets2 (ETSD) shares extremely high sequence similarity to the Ets domain of Ets1 (about 96%). Ets1 is one of the most structurally investigated Ets-family members. The crystal structures of Ets1(331–440) (the Ets domain and its C-terminal inhibitory region) complexed with Pax5(1–149) and DNA, with high-affinity type core GGAA DNA and with low-affinity type core GGAG DNA, of Ets1(301–440) (the Ets domain and its N- and C-terminal inhibitory regions) with Pax5(1–149) and DNA and of Ets1(281–440) have been determined. Comparison of Ets1(331–440)–Pax5–DNA with Ets1(331–440)–DNA reveals that the direct interaction between Pax5 and the Ets1 key recognition residue Tyr395Ets1 alters the side-chain conformation of Tyr395Ets1 and facilitates the binding of Ets1 to a low-affinity DNA sequence. The structures of Ets1(301–440) and Ets1(281–440)–Pax5–DNA indicate that the N-terminal inhibitory region of the Ets domain contains two α-helices, one of which unfolds upon the binding of Ets1 to DNA. As mentioned above, Ets2 has a similar Ets domain to Ets1; however, their biological functions are unique. Furthermore, Ets2 is ubiquitously expressed in many cell lines and is co-expressed with Ets1 in most cell lines (Hollenhorst et al., 2004 ▶). This extensive co-expression, in addition to the conservation of the Ets domain, provides a challenge to the possession of in vivo specificity. In general, the DNA-binding affinities between Ets2 and DNA and protein–protein interactions between Ets2 and other factors may play a key role in specific transcriptions. To elucidate the structural basis of the specific biological functions of Ets2, the structures of ETSD(359–446)–DNA, Ets2(308–470)–DNA, Ets2(308–470) and Ets2(1–470)–DNA and their complexes with interaction partners are required. In this study, we focus on the structure of ETSD(359–446) in complex with its target DNA in order to understand the innate preference of ETSD for Ets-binding site sequences on promoters and its differences from the structures of previously determined Ets1 crystals. Usually, Ets domains are liable to aggregation (Pio et al., 1995 ▶). We succeeded in crystallizing the ETSD–DNA complex by a novel method using NDSB-195 to increase protein solubility.
2. Experimental procedures and results
2.1. Protein expression and purification
The gene for ETSD(359–446) was amplified by the polymerase chain reaction with Pyrobest polymerase (TaKaRa) using full-length Ets2 cDNA (kindly provided by Dr D. K. Watson) as the template. The PCR product was subcloned into a pGEX-1λT vector (GE Healthcare Biosciences) using a BamHI/EcoRI restriction enzyme (TakaRa) site. The N-terminal GST-fusion expression plasmid pGEX-1λT/ETSD was transformed into Escherichia coli strain BL21(DE3)RIL and the cells were grown at 310 K in Luria–Bertani broth. When the cells reached an OD660 of 0.6, isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 0.1 mM. After cell growth for 5 h at 298 K, the cells were harvested at 277 K, resuspended in 50 mM sodium phosphate pH 7.0, 500 mM NaCl, 2 mM 2-mercaptoethanol and 1 mM EDTA and lysed by sonication on ice. The fusion protein GST-ETSD was initially purified by applying the supernatant from the cell lysate onto a glutathione-affinity column. The elution fractions containing GST-ETSD were dialyzed against 50 mM Tris–HCl pH 8.0, 500 mM NaCl, 2 mM 2-mercaptoethanol and 1 mM EDTA to remove glutathione prior to thrombin digestion. When GST-ETSD was treated with thrombin at 293 K, the proteolyzed form of ETSD (including two additional residues, Gly and Ser, derived from the expression-vector sequence at its N-terminus) immediately precipitated. The precipitation of ETSD could be prevented by the addition of NDSB-195 (Merck) to the digestion solution at a final concentration of 1 M (Blisnick et al., 1998 ▶; Chong & Chen, 2000 ▶; Expert-Bezancon et al., 2003 ▶; Vuillard et al., 1994 ▶, 1995 ▶). In this digestion condition with NDSB-195, a fivefold amount of thrombin was required compared with the usual treatment in order to cleave the fusion protein. After thrombin treatment for 18 h at 293 K, the solution was diluted to 300 mM NaCl with 50 mM Tris–HCl pH 8.0, 2 mM 2-mercaptoethanol and 1 mM EDTA. ETSD in the diluted solution was purified by heparin-affinity and cation-exchange column chromatography.
2.2. Cocrystallization
The oligonucleotides used for crystallization were obtained from Hokkaido System Sciences (Sapporo, Japan). All of the oligonucleotides contained the Ets2 target sequence, GGAA, and some were designed to have a one-base overhang at their 5′ ends in order to facilitate base stacking and pairing between neighbouring molecules in the crystal lattice. Equimolar amounts of the complementary strands were mixed in TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA). The mixture was heated to 371 K for 5 min and then cooled slowly to room temperature. Double-stranded DNA (dsDNA) formation was verified by 12% native PAGE analysis.
After cation-exchange column chromatography, purified ETSD in 50 mM Tris–HCl pH 8.0, 1 M NaCl, 1 mM EDTA and 2 mM 2-mercaptoethanol was mixed with a variety of dsDNA molecules in a 1:1.5 molar ratio of ETSD to dsDNA and dialyzed against 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM EDTA and 2 mM 2-mercaptoethanol to form the ETSD–DNA complex. Complex formation was confirmed by running a 12% native PAGE and staining with Coomassie Brilliant Blue (CBB) and ethidium bromide (EtBr). These complex solutions were concentrated to approximately 3.0 mg ml−1 and used for initial crystallization screening at 293 K utilizing commercially available screening kits such as Natrix, Crystal Screen, Crystal Screen II (Hampton Research), Wizard I, Wizard II, Wizard III and Precipitant Synergy Screen (Emerald Biosystem) by the hanging-drop vapour-diffusion method. 1 µl complex solution was mixed with an equal volume of reservoir solution and the mixture was equilibrated against 0.4 ml reservoir solution. In the crystallization of protein–DNA complexes, dsDNA with various lengths (11–16 bases), configurations (one-base 5′ overhang and blunt-end) and base sequences at the 5′ and 3′ ends (A, C, G or T; Pio et al., 1995 ▶) were screened. According to the screening results, DNA with 15 base pairs and a one-base overhang at the 5′ end of each strand (5′-AAAGTGCCGGAAATGT-3′ and 5′-TACATTTCCGGCACTT-3′) was used for subsequent crystallizations. Based on a condition that produced single crystals, the pH and the concentrations of metal ions and precipitants were modified. Crystals of suitable dimensions for X-ray diffraction experiments were grown when 3 µl complex solution containing 3.0 mg ml−1 complex, 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM EDTA and 2 mM 2-mercaptoethanol was mixed with 3 µl 18% PEG 4000, 80 mM magnesium acetate and 50 mM sodium cacodylate pH 5.0/5.5. However, the crystals only diffracted to about 7 Å resolution. Therefore, the ETSD–DNA complex was further purified by gel-filtration column chromatography. The formation of the ETSD–DNA complex and the purity of the purified ETSD after gel filtration were evaluated by 12% native PAGE and 17% SDS–PAGE (Fig. 1 ▶). Subsequent optimization of the PEG molecular weight and concentration using PEG 400, 1000, 3350, 4000, 6000 and 8000 resulted in single crystals that were suitable for crystallographic experiments. The crystals grew after 7–14 d at 293 K on mixing 1 µl ETSD–DNA solution containing 3.0 mg ml−1 complex, 50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM EDTA and 2 mM 2-mercaptoethanol with 1 µl 25% PEG 3350, 80 mM magnesium acetate and 50 mM sodium cacodylate pH 5.0/5.5 (Fig. 2 ▶).
Figure 1.

PAGE analyses of the ETSD–DNA complex after gel-filtration column chromatography. (a) 12% native PAGE gel stained with CBB (upper) and then with EtBr (lower); lane 1, pre-gel filtration sample without membrane filtration; lane 2, pre-gel filtration sample after membrane filtration; lanes 3–11, fractions eluted from gel filtration. (b) 17% SDS–PAGE gel of the purified ETSD–DNA complex used for crystallization stained with CBB; lane 1, molecular-weight markers (kDa); lane 2, crystallization sample.
Figure 2.

Typical crystals of the ETSD–DNA complex. The maximum dimensions of the crystals are 0.2 × 0.4 × 0.05 mm.
2.3. Data collection and processing
All crystals were transferred into a cryoprotectant composed of 20%(v/v) glycerol, 25% PEG 3350, 80 mM magnesium acetate and 50 mM sodium cacodylate pH 5.0/5.5, mounted in a cryoloop (Hampton Research) and flash-frozen in a nitrogen-gas stream at 100 K. Diffraction data were collected on beamlines BL44XU and BL41XU at SPring-8 (Harima, Japan) and BL5A and NW12 at Photon Factory (Tsukuba, Japan) at 100 K. Although the best data set at 100 K was collected to a resolution of 3.0 Å, strong anisotropy of the data was observed. We therefore tried to collect data at 40 K using a helium-gas stream at SPring-8 and obtained an improved data set (Fig. 3 ▶). All data were processed, integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶). The ETSD–DNA complex crystals belong to space group C2, with unit-cell parameters a = 85.89, b = 95.52, c = 71.89 Å, β = 101.7°. Assuming the presence of two ETSD–DNA complexes per asymmetric unit, the V M value is 3.56 Å3 Da−1 and the calculated solvent content is 65% (Matthews, 1968 ▶). The data-collection statistics of the data set used for structural determination are presented in Table 1 ▶.
Figure 3.

X-ray diffraction image for the ETSD–DNA complex.
Table 1. Data-collection statistics.
Values in parentheses correspond to the highest resolution shell.
| Beamline | BL41XU, SPring-8 |
| Wavelength (Å) | 1.0 |
| Detector | ADSC Quantum 315 CCD |
| Exposure time (s) | 3.0 |
| Crystal-to-detector distance (mm) | 370 |
| Oscillation angle (°) | 1.0 |
| Sweep angle (°) | 0–360 |
| Temperature (K) | 40 |
| Crystal data | |
| Space group | C2 |
| Unit-cell parameters | |
| a (Å) | 85.89 |
| b (Å) | 95.52 |
| c (Å) | 71.89 |
| β (°) | 101.7 |
| Unit-cell volume (Å3) | 577500 |
| VM (Å3 Da−1) | 3.56 |
| Solvent content (%) | 65 |
| No. of molecules per ASU | 2 ETSD–DNA complexes |
| Data statistics | |
| Resolution (Å) | 50.0–3.0 (3.11–3.0) |
| No. of observed reflections | 70267 (4172) |
| No. of unique reflections | 11042 (927) |
| Redundancy | 6.4 (4.5) |
| Completeness (%) | 95.4 (76.2) |
| 〈I/σ(I)〉 | 34.9 (3.58) |
| Rmerge† | 0.088 (0.413) |
R
merge = 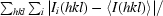
 , where I(hkl) is the observed intensity and 〈I(hkl)〉 is the mean value of I(hkl).
, where I(hkl) is the observed intensity and 〈I(hkl)〉 is the mean value of I(hkl).
2.4. Crystallographic analysis
The structure of the ETSD–DNA complex was solved by molecular replacement to 4.0 Å resolution using the program MOLREP (Collaborative Computational Project, Number 4, 1994 ▶; Vagin & Teplyakov, 2000 ▶) with the Ets1(331–440)–DNA complex structure (PDB code 1k79; Garvie et al., 2001 ▶) as a search model, with protein residues 333–415 corresponding to 361–443 of Ets2 and a 13 base-pair DNA duplex. A single solution with a correlation coefficient of 0.53 and an R factor of 0.47 was obtained after the translation-function calculation considering two independent protein–DNA complexes. The next best solution had a correlation coefficient of 0.31 and an R factor of 0.56. The initial 2F o − F c map calculated at 3.0 Å resolution showed unambiguous density for the remaining 12 nucleotides (two 5′-end and one 3′-end nucleotides in each DNA strand; four strands per asymmetric unit). The current structure model reveals that a pseudo-continuous double helix is formed by two independent DNA duplexes with an AT base pair between the one-base 5′ overhangs of each strand and by the same AT base pair in the crystallographically related neighbours. Structure refinement is in progress.
Acknowledgments
We thank Dr D. K. Watson for kindly providing the human Ets2 cDNA and Dr M. A. Suico for critical reading of the manuscript. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Sciences and Technology of Japan.
References
- Baldus, C. D., Liyanarachchi, S., Mrózek, K., Auer, H., Tanner, S. M., Guimond, M., Ruppert, A. S., Mohamed, N., Davuluri, R. V., Caligiuri, M. A., Bloomfield, C. D. & de la Chapelle, A. (2004). Proc. Natl Acad. Sci. USA, 101, 3915–3920. [DOI] [PMC free article] [PubMed]
- Blisnick, T., Morales-Betoulle, M. E., Vuillard, L., Rabilloud, T. & Braun Breton, C. (1998). Eur. J. Biochem.252, 537–541. [DOI] [PubMed]
- Chong, Y. & Chen, H. (2000). Biotechniques, 29, 1166–1167. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Dwyer, J., Li, H., Xu, D. & Liu, J. P. (2007). Ann. NY Acad. Sci.1114, 36–47. [DOI] [PubMed]
- Escalante, C. R., Brass, A. L., Pongubala, J. M., Shatova, E., Shen, L., Singh, H. & Aggarwal, A. K. (2002). Mol. Cell, 10, 1097–1105. [DOI] [PubMed]
- Expert-Bezancon, N., Rabilloud, T., Vuillard, L. & Goldberg, M. E. (2003). Biophys. Chem.100, 469–479. [DOI] [PubMed]
- Foulds, C. E., Nelson, M. L., Blaszczak, A. G. & Graves, B. J. (2004). Mol. Cell. Biol.24, 10954–10964. [DOI] [PMC free article] [PubMed]
- Garvie, C. W., Hagman, J. & Wolberger, C. (2001). Mol. Cell, 8, 1267–|1276. [DOI] [PubMed]
- Garvie, C. W., Pufall, M. A., Graves, B. J. & Wolberger, C. (2002). J. Biol. Chem.277, 45529–45536. [DOI] [PubMed]
- Garvie, C. W. & Wolberger, C. (2001). Mol. Cell, 8, 937–946. [DOI] [PubMed]
- Hollenhorst, P. C., Jones, D. A. & Graves, B. J. (2004). Nucleic Acids Res.32, 5693–5702. [DOI] [PMC free article] [PubMed]
- Hollenhorst, P. C., Shah, A. A., Hopkins, C. & Graves, B. J. (2007). Genes Dev.21, 1882–1894. [DOI] [PMC free article] [PubMed]
- Hsu, C. P., Hsu, N. Y., Lee, L. W. & Ko, J. L. (2006). Eur. J. Cancer, 42, 1466–1474. [DOI] [PubMed]
- Kodandapani, R., Pio, F., Ni, C. Z., Piccialli, G., Klemsz, M., McKercher, S., Maki, R. A. & Ely, K. R. (1996). Nature (London), 380, 456–460. [DOI] [PubMed]
- Li, M., Zhang, Z., Hill, D. L., Wang, H. & Zhang, R. (2007). Cancer Res.67, 1988–1996. [DOI] [PubMed]
- Li, R., Pei, H. & Watson, D. K. (2000). Oncogene, 19, 6514–6523. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mo, Y., Vaessen, B., Johnston, K. & Marmorstein, R. (1998). Mol. Cell, 2, 201–212. [DOI] [PubMed]
- Mo, Y., Vaessen, B., Johnston, K. & Marmorstein, R. (2000). Nature Struct. Biol.7, 292–297. [DOI] [PubMed]
- Obika, S., Reddy, S. Y. & Bruice, T. C. (2003). J. Mol. Biol.331, 345–359. [DOI] [PubMed]
- Oikawa, T. & Yamada, T. (2003). Gene, 303, 11–34. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pio, F., Kodandapani, R., Ni, C. Z., Shepard, W., Klemsz, M., McKercher, S. R., Maki, R. A. & Ely, K. R. (1996). J. Biol. Chem.271, 23329–23337. [DOI] [PubMed]
- Pio, F., Ni, C. Z., Mitchell, R. S., Knight, J., McKercher, S., Klemsz, M., Lombardo, A., Maki, R. A. & Ely, K. R. (1995). J. Biol. Chem.270, 24258–24263. [DOI] [PubMed]
- Raouf, A. & Seth, A. (2000). Oncogene, 19, 6455–6463. [DOI] [PubMed]
- Sacchi, N., Cheng, S. V., Tanzi, R. E., Gusella, J. F., Drabkin, H. A., Patterson, D., Haines, J. H. & Papas, T. S. (1988). Genomics, 3, 110–116. [DOI] [PubMed]
- Sementchenko, V. I. & Watson, D. K. (2000). Oncogene, 19, 6533–6548. [DOI] [PubMed]
- Sharrocks, A. D. (2001). Nature Rev. Mol. Cell Biol.2, 827–837. [DOI] [PubMed]
- Tynan, J. A., Wen, F., Muller, W. J. & Oshima, R. G. (2005). Oncogene, 24, 6870–6876. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2000). Acta Cryst. D56, 1622–1624. [DOI] [PubMed]
- Vuillard, L., Braun-Breton, C. & Rabilloud, T. (1995). Biochem. J.305, 337–343. [DOI] [PMC free article] [PubMed]
- Vuillard, L., Rabilloud, T., Leberman, R., Berthet-Colominas, C. & Cusack, S. (1994). FEBS Lett.353, 294–296. [DOI] [PubMed]
- Wang, Y., Feng, L., Said, M., Balderman, S., Fayazi, Z., Liu, Y., Ghosh, D. & Gulick, A. M. (2005). Biochemistry, 44, 7095–7106. [DOI] [PubMed]
- Watson, D. K., McWilliams-Smith, M. J., Nunn, M. F., Duesberg, P. H., O’Brien, S. J. & Papas, T. S. (1985). Proc. Natl Acad. Sci. USA, 82, 7294–7298. [DOI] [PMC free article] [PubMed]
- Wolvetang, E. J., Bradfield, O. M., Hatzistavrou, T., Crack, P. J., Busciglio, J., Kola, I. & Hertzog, P. J. (2003). Neurobiol. Dis.14, 349–356. [DOI] [PubMed]
- Wolvetang, E. J., Wilson, T. J., Sanij, E., Busciglio, J., Hatzistavrou, T., Seth, A., Hertzog, P. J. & Kola, I. (2003). Hum. Mol. Genet.12, 247–255. [DOI] [PubMed]
- Yang, B. S., Hauser, C. A., Henkel, G., Colman, M. S., Van Beveren, C., Stacey, K. J., Hume, D. A., Maki, R. A. & Ostrowski, M. C. (1996). Mol. Cell. Biol.16, 538–547. [DOI] [PMC free article] [PubMed]