

The crystal structure of ADP-ribosylhydrolase 3 from M. musculus has been determined and refined to a resolution of 1.8 Å. A detailed comparison with the human orthologue at the protein-sequence level as well as of the three-dimensional architecture is presented.
Keywords: ADP-ribosylhydrolase 3
Abstract
ADP-ribosylation is a reversible and covalent post-translational modification in which the attachment of ADP-ribose is catalyzed by ADP-ribosyltransferases and the removal of ADP-ribose is catalyzed by ADP-ribosylhydrolases. ADP-ribosylhydrolase 3 from mouse, consisting of 347 amino-acid residues, has been cloned, purified and crystallized. The three-dimensional structure has been resolved at a resolution of 1.8 Å. The structure constitutes a compact all-α-helical protein with two Mg2+ ions located in the active-site crevice. A structural comparison of mouse ADP-ribosylhydrolase 3 with its human orthologue shows a high degree of structural similarity. Furthermore, four prokaryotic proteins deposited in the PDB could be identified as being structurally related.
1. Introduction
The addition of ADP-ribose onto proteins is a reversible post-translational modification that is catalysed by a variety of enzymes (Shall, 1995 ▶). It can occur either in the form of mono-ADP-ribosylation (Seman et al., 2004 ▶) or in the form of poly-ADP-ribosylation (Hassa et al., 2006 ▶). ADP-ribosylation is the reaction in which the ADP-ribose moiety of NAD is transferred to a specific acceptor amino-acid residue such as Arg, Asn, Glu, Asp, Cys, diphthamide or phosphoserine. During the stereospecific chemical reaction, the target performs a nucleophilic attack on the anomeric C atom of the ribose moiety adjacent to nicotinamide in NAD, thereby generating a covalent linkage with ADP-ribose and releasing nicotinamide. The hydrolysis of NAD itself can be described as a transfer of ADP-ribose onto a water molecule.
Many of the bacterial mono-ADP-ribosyltransferases (mARTs) are known for their toxicity. Prominent examples are diphtheria toxin, cholera toxin, Salmonella virulence factor SpvB and many others (Aktories & Just, 2000 ▶). ADP-ribosylating toxins catalyse the transfer of the ADP-ribose moiety from NAD to specific target proteins, in many cases GTP-binding or ATP-binding proteins. ADP-ribosylation often alters the function of the target protein and disrupts cellular metabolism. SpvB-related mammalian ecto-mARTs occur as glycosylphosphatidylinositol-anchored or secretory proteins that can be divided into five paralogues: mART1–5 (Glowacki et al., 2002 ▶; Seman et al., 2004 ▶). Diphtheria-toxin-related poly-ADP-ribosyltransferases or poly-(ADP-ribose) polymerases (pARTs or PARPs) are intracellular, usually nuclear, proteins of eukaryotes (Otto et al., 2005 ▶; Schreiber et al., 2006 ▶) that play a role in DNA repair (Malanga & Althaus, 2005 ▶), transcriptional activation and repression (Kim et al., 2004 ▶), chromatin insulation (Yu et al., 2004 ▶), cell death (Jagtap & Szabo, 2005 ▶), regulation of mitosis (Chang et al., 2004 ▶) or the regulation of telomere length (Smith et al., 1998 ▶). It has been suggested that some members of the mART and PARP families can act both as a mART and as a PARP (Otto et al., 2005 ▶; Hassa et al., 2006 ▶; Morrison et al., 2006 ▶). Additionally, there is evidence for intracellular ADP-ribosylation, which may be catalyzed by certain members of the structurally distinct sirtuin family. For instance, SIRT4 has been shown to regulate insulin secretion by mono-ADP-ribosylating glutamate dehydrogenase (Haigis et al., 2006 ▶; Ahuja et al., 2007 ▶).
The extent and duration of mono-ADP-ribosylation depends on the activity of ADP-ribosylhydrolases (ARHs) and poly-ADP-ribosylglycohydrolases (PARGs), which reverse the transfer reaction by hydrolysing the protein–ADP-ribose bond and/or ADP-ribose–ADP-ribose bonds (Koch-Nolte & Haag, 1997 ▶; Moss et al., 1997 ▶). In this context, mARTs/PARPs and ARHs/PARGs form opposite pairs in the ADP-ribosylation reaction cycle (Fig. 1 ▶). To date, three mammalian ARHs (ARH1–3) have been described (Glowacki et al., 2002 ▶), where ARH1 has been reported to cleave ADP-ribosyl-arginine (Moss et al., 1997 ▶) and ARH3 to de-ADP-ribosylate poly-ADP-ribosylated proteins (Oka et al., 2006 ▶; Mueller-Dieckmann et al., 2006 ▶) and to hydrolyse O-acetyl-ADP-ribose (Ono et al., 2006 ▶). ARH3 localizes to the mitochondrial matrix, where it can efficiently degrade poly-ADP-ribose generated by mitochondrially targeted PARP1 in living cells (Niere et al., 2008 ▶). PARG is encoded by a single-copy gene in mammals, which is transcribed into different splice variants that mediate differential subcellular localization to the nucleus, cytosol and mitochondria (Meyer et al., 2007 ▶). PARG cleaves ADP-ribosyl-ADP-ribose linkages but is not known to cleave ADP-ribosyl-amino-acid linkages.
Figure 1.

The reversible ADP-ribosylation reaction. ARTs and PARPs catalyse the transfer of an ADP-ribose moiety from NAD onto specific amino-acid residues or onto ADP-ribose, respectively, thereby releasing nicotinamide. The reverse reaction is catalysed by ARHs or PARGs through the hydrolysis of the α-glycosidic bond between ADP-ribose and the side chain. X can be Arg, Asp, Glu, Asn, Cys, diphthamide or ADP-ribose. For mono-ADP-ribosylation R and R′ are hydroxyl groups, while for poly-ADP-ribosylation ADP-ribose can be attached to the R site (elongation) or to the R′ site (branching).
The only structurally characterized member of the ARH and PARG protein families is human ARH3 (Kernstock et al., 2006 ▶; Mueller-Dieckmann et al., 2006 ▶). The crystal structure of human ARH3 revealed two Mg2+ ions to be localized in the active-site crevice. Since these are essential for the full activity of the protein, which has been demonstrated for the related enzyme ARH1 (Moss et al., 1985 ▶), they pinpoint the active site. Binding of ADP-ribose to human ARH3 has been confirmed by isothermal titration microcalorimetry experiments and the binding mode was evaluated by docking studies (Mueller-Dieckmann et al., 2006 ▶). Site-directed mutagenesis of amino-acid residues predicted to be involved in binding ADP-ribose and/or the magnesium ions resulted in a severe loss of enzymatic activity, supporting the suggested binding mode (Mueller-Dieckmann et al., 2006 ▶).
Mouse ARH3 (ADPRHL2) consists of 370 amino acids and shares 17 and 16% amino-acid sequence identity with the mouse paralogues ARH1 and ARH2, respectively, and 90% identity with the human orthologue. Here, we present the structure of ARH3 from Mus musculus refined to a resolution of 1.8 Å and a comparison with its human orthologue and with other structurally related proteins.
2. Materials and methods
2.1. Cloning, expression and purification
A cDNA clone encompassing the coding region for mARH3 was obtained from the IMAGE consortium (IMAGE Consortium Clone ID 5293159; Lennon et al., 1996 ▶). The ARH coding region excluding a putative N-mitochondrial targeting sequence (residues 2–23) was amplified by PCR with appropriate primers (5′-CGATGACCATATGCGCTCCATCTCGCGCTTCCG-3′ and 5′-AAGCACTCGAGCGAGCTCTCCTGGAAGACTCGGT-3′) and cloned into the NdeI and XhoI sites of the prokaryotic expression plasmid pET26b (Novagen). This cloning strategy resulted in the removal of the pelB leader sequence from pET26b, the deletion of residues 2–23 of mARH3 and a C-terminal extension of two amino acids encoded by the XhoI recognition sequence (Leu-Glu) followed by a hexahistidine tag. The correct cloning of the gene was verified by DNA sequencing.
The expression plasmid pET26b-mARH3 was freshly transformed into Escherichia coli BL21-CodonPlus(DE3)-RIL cells (Stratagene). A single colony was picked for an overnight preculture, which took place at 310 K in LB broth medium containing 50 µg ml−1 kanamycin and 30 µg ml−1 chloramphenicol. 5–10 ml of this preculture was used to inoculate a 1 l culture, which was grown at 310 K to an optical density at 600 nm (OD600) of approximately 0.5. The temperature was then reduced to 293 K and expression was induced by adding isopropyl β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM when an OD600 of about 0.7 was reached. After induction, cells were grown for an additional 15 h and harvested. The cells were then frozen at 253 K until further processing.
1 g of the cells was suspended in 4 ml lysis buffer [50 mM Tris pH 8.0, 150 mM NaCl, 3 mM β-mercaptoethanol (β-ME), 10 mM MgCl2 and 0.1%(v/v) Trition X-100]. After lysis by sonication, the cell debris was removed by centrifugation at 20 000 rev min−1 at 277 K for 30 min. The clear supernatant was passed onto an Ni–NTA Superflow column (Qiagen) that had been equilibrated with lysis buffer. After extensive washing (ten column volumes) with washing buffer (50 mM Tris pH 8.0, 300 mM NaCl, 3 mM β-ME and 5 mM MgCl2), the protein was eluted in a buffer containing 50 mM Tris pH 8.0, 300 mM NaCl, 3 mM β-ME, 5 mM MgCl2 and 500 mM imidazole. The eluate was concentrated to a volume of less than 1 ml and directly applied onto a Sephacryl-S200 size-exclusion column (Amersham Pharmacia). mARH3 eluted in a buffer containing 50 mM Tris pH 8.0, 150 mM NaCl and 2 mM DTT with a molecular weight of about 40 000 Da, corresponding to a monomer in solution. Protein-containing fractions exhibiting a purity of higher than 95% were pooled, concentrated and dialyzed extensively against 10 mM HEPES pH 7.0 and 150 mM NaCl. Typical yields were around 15 mg of pure protein per litre of culture.
2.2. Crystallization
Prior to crystallization, the protein concentration was adjusted to 5 mg ml−1. Initial screening for crystals was performed at the High-Throughput Crystallization facility of the EMBL Outstation in Hamburg (Mueller-Dieckmann, 2006 ▶). The sample was tested in five commercial initial screens (Crystal Screens I and II, Index and SaltRx from Hampton Research and Jena 1–8 from Jena Bioscience). In the refined crystallization conditions, crystals were grown at 295 K, mixing protein and reservoir solution [100 mM MES pH 6.2 and 6%(w/v) PEG 4000] in a 1:1 ratio. Crystals appeared within one to two weeks as long needles that were often badly grown together (up to 1 mm in length and 150 µm in width; Fig. 2 ▶).
Figure 2.

Crystal of mARH3. The bar in the upper right corner corresponds to 100 µm.
2.3. Structure determination and refinement
Prior to diffraction data collection, a crystal of mARH3 was cryoprotected by soaking it in 20%(v/v) glycerol in reservoir solution for 20 s and then flash-cooled in a 100 K nitrogen stream. Diffraction data were then collected to a diffraction limit of 1.8 Å (Table 1 ▶) at EMBL beamline X11 (DESY Hamburg, Germany) using a MAR CCD (165 mm) detector. The data were indexed, integrated and scaled using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). Intensities were converted to structure-factor amplitudes using the program TRUNCATE (French & Wilson, 1978 ▶; Collaborative Computational Project, Number 4, 1994 ▶). Initial phases were obtained by molecular replacement with the program MOLREP (Vagin & Teplyakov, 1997 ▶; Collaborative Computational Project, Number 4, 1994 ▶) using the human orthologue (Mueller-Dieckmann et al., 2006 ▶) as a search model (PDB code 2foz). After having correctly oriented and placed both molecules in the asymmetric unit, the model amino-acid sequence was modified to reflect the sequence of mouse ARH3 using the program Coot (Emsley & Cowtan, 2004 ▶; Collaborative Computational Project, Number 4, 1994 ▶). Refinement was performed using the program REFMAC5 (Winn et al., 2001 ▶; Collaborative Computational Project, Number 4, 1994 ▶). Refinement statistics are summarized in Table 2 ▶.
Table 1. Data-collection and processing statistics.
| Data-collection wavelength | 0.8126 |
| Space group | P21 |
| Unit-cell parameters | |
| a (Å) | 53.88 |
| b (Å) | 60.10 |
| c (Å) | 91.78 |
| β (°) | 90.09 |
| Resolution limits (Å) | 99–1.80 (1.83–1.80) |
| Mosaicity (°) | 1.0 |
| Total no. of reflections | 345718 |
| No. of unique reflections | 51153 |
| Redundancy | 6.8 (6.5) |
| 〈I/σ(I)〉 | 23.9 (2.5) |
| Completeness | 93.8 (89.2) |
| Rmerge† (%) | 7.7 (80.3) |
| Rr.i.m.‡ (%) | 8.3 (87.0) |
| Rp.i.m.‡ (%) | 3.1 (32.9) |
| Optical resolution (Å) | 1.49 |
| Overall B factor from Wilson plot (Å2) | 24.8 |
R
merge(I) = 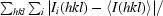
 .
.
The redundancy-independent merging R factor (R
r.i.m. = 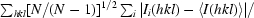 ) and the precision-indicating merging R factor {R
p.i.m. =
) and the precision-indicating merging R factor {R
p.i.m. = 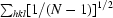
 } (Weiss, 2001 ▶) were calculated using the program RMERGE (available from MSW upon request or from http://www.embl-hamburg.de/~msweiss/projects/msw_qual.html).
} (Weiss, 2001 ▶) were calculated using the program RMERGE (available from MSW upon request or from http://www.embl-hamburg.de/~msweiss/projects/msw_qual.html).
Table 2. Refinement statistics.
| Refinement | |
| Resolution limits (Å) | 30–1.80 |
| No. of reflections | |
| Working set | 50111 |
| Test set | 975 |
| Rcryst (%) | 16.7 |
| Rfree (%) | 19.8 |
| No. of atoms | |
| Protein | 5181 |
| Ions | 4 |
| Waters | 200 |
| Stereochemistry | |
| R.m.s. deviations | |
| Bonds (Å) | 0.016 |
| Angles (°) | 1.47 |
| Average B factors (Å2) | |
| Protein | 18.3 |
| Ions | 12.5 |
| Waters | 19.6 |
| Ramachandran plot | |
| Most favoured (%) | 97.0 |
| Allowed (%) | 2.4 |
3. Results and discussion
3.1. Overall structure
The complete model consists of 674 amino acids in two protomers, four Mg2+ ions and 200 water molecules. Although the content of the asymmetric unit of the crystal is two protomers, the most likely biological unit is a monomer, as also evidenced by size-exclusion chromatography, which indicates that the protein is monomeric in solution. Both all-α-helical protomers are composed of 19 α-helices, with two Mg2+ ions that pinpoint the active-site crevice, and have dimensions of approximately 53 × 50 × 62 Å3 (Fig. 3 ▶ and Fig. 4 ▶). The first 23 N-terminal amino-acid residues, i.e. the predicted mitochondrial targeting sequence, have been replaced by a Met residue during cloning. The loop regions from Asp45 to Thr54 in both protomers A and B as well as the two C-terminal serine residues and the subsequent His6 tag are missing from the model owing to ill-defined electron density. The two protomers in the asymmetric unit exhibit an average r.m.s. deviation of only 0.69 Å for all 335 superimposed Cα positions. A comparison of the B-factor distribution with the difference of Cα positions in both protomers shows that the regions exhibiting higher B factors generally also exhibit the highest Cα discrepancies. The four internal loop regions are (i) the ‘missing’ loop (residues 45–54), (ii) the loop region between α-helices 10 and 11 (residues 188–192), which is situated at the crystal contact between the two protomers, (iii) the loop region between α-helices 12 and 13 (residues 218–224), which is also on the surface, and (iv) the loop between α-helices 15 and 16 (residues 271–283), which like (ii) can be found at the crystal contact of the two protomers.
Figure 3.

Ribbon-plot representation of mARH3. The colouring scheme is from the N-terminus (blue) to the C-terminus (red). It is identical to that chosen for the human orthologue (Mueller-Dieckmann et al., 2006 ▶) for better comparison. The two magnesium ions in the active-site crevice are shown as cyan spheres. The ill-defined loop between α-helices 2 (dark blue) and 3 (light blue) has not been drawn in the representation.
Figure 4.

Ribbon-plot representation and protein solvent-accessible surface representation. The colouring scheme for the protein chain is from the N-terminus (blue) to the C-terminus (red). The solvent-accessible surface is coloured grey and the two Mg2+ ions are shown as cyan spheres.
3.2. Active site
The chain termini are situated close to each other on the same side of the molecule. Two Mg2+ ions are located at the opposite side of the molecule within a cleft that spans nearly the entire width of the molecule. These metal ions are separated from each other by 3.74 Å and pinpoint the active site in which the ADP-ribose polymer is hydrolysed. The four tightly bound ions (two per protomer) show an average B factor of 12.5 Å2, which is lower than the average B factor for the protein itself (18.3 Å2) and explains why Mg2+ ions did not have to be present during size-exclusion chromatography and crystallization. Each of the Mg2+ ions is nearly perfectly octahedrally coordinated by three and five acidic residues (Glu and Asp), respectively, one Thr residue and two water molecules. The corresponding residues, distances and deviation from ideal geometry are listed in Table 3 ▶. While the distances to the coordinating atoms for the first Mg2+ ion deviate on average just 4.5% from ideal distances, the deviation from ideality is much higher for the second Mg2+ ion (16.9%).This is accompanied by a higher flexibility for the second ion (B factor of 12.9 Å2 compared with 8.2 Å2 for the first ion). It is this tempting to speculate that the first Mg2+ ion serves a structural role while the second Mg2+ ion contributes to enzymatic activity by acting as an electron sink. This notion is corroborated by docking experiments performed on human ARH3, in which the second Mg2+ ion coordinates the 2′-hydroxyl group of the ribose carrying the scissile glycosidic bond (Mueller-Dieckmann et al., 2006 ▶).
Table 3. Coordination of the two Mg2+ ions.
Bridging atoms are highlighted in bold. Mean distances from structures in the PDB at near-atomic resolution are 2.09 Å for Mg2+—OH2, 2.10 Å for Mg2+—O Thr, 2.08 Å for monodentate Mg2+—−OOC Asp/Glu and 2.5 Å as the maximal accepted distance of bidentate Mg2+—−OOC Asp/Glu (Harding, 2001 ▶, 2006 ▶). The two coordinating atoms of the Mg-II ion that differ by more than 50% from the mean distance are indicated in italics. Values for the deviation of the mean distance assuming a bidentate coordinating nature are given in parentheses.
| Coordinating atom | Distance (Å) | Deviation from mean distance (%) |
|---|---|---|
| Mg-I | ||
| Thr60 OG1 | 2.32 | +10.5 |
| Asp61 OD1 | 2.03 | −2.4 |
| Asp62 OD2 | 1.93 | −7.2 |
| Asp300 OD2 | 2.01 | −3.4 (19.6) |
| W20 | 2.10 | +0.5 |
| W61 | 2.02 | −3.3 |
| Mg-II | ||
| Glu25 OE2 | 2.26 | +8.7 |
| Asp298 OD1 | 2.17 | +4.3 (13.2) |
| Asp298 OD2 | 3.42 | +64.4 (36.8) |
| Asp300 OD1 | 2.20 | +5.8 (12.0) |
| Asp300 OD2 | 3.16 | +51.2 (26.4) |
| Thr301 OG1 | 2.32 | +10.5 |
| W38 | 2.27 | +8.6 |
| W61 | 2.23 | +6.7 |
3.3. Comparison with human ADP-ribosylhydrolase 3
A superposition of the two protomers of ARH3 from mouse with the structure of human ARH3 (PDB code 2foz) shows an r.m.s. deviation of only 0.40 Å for 336 superimposed Cα atoms for the first protomer and 0.52 Å for 331 superimposed Cα atoms for the second protomer. Compared with the human ARH3 protein, there are a total of 26 substituted amino-acid residues, of which 17 can be described as conservative substitutions (Fig. 5 ▶). Table 4 ▶ lists all of the substitutions between mouse and human ARH3. All but two substitutions are located on the protein surface and do not alter the electrostatic surface potential. The two substitutions which are not located on the protein surface but in the protein core are Thr66 and Val233, which are Ala and Ile in the human orthologue, respectively. Both substitutions are far away from the active-site crevice. Thr66 is located on α-helix 3 and its closest distance to the Mg2+ ions is around 10 Å. It forms a hydrogen bond to the carbonyl O atom of Asp62 through its hydroxyl group (2.8 Å), which is in turn involved in the Mg2+ coordination. The substitution of Ala by Thr increases the number of hydrogen bonds formed by the carbonyl O atom of Asp62 from three to four. A comparison with the human protein shows that the position of Asp62 is unaltered, suggesting that the substitution has no effect on the activity of the protein. Val233, on the other hand, is located around 19 Å away from the active site on α-helix 12 at the hydrophobic interface with α-helix 14. The substitution of Ile by Val leaves a hole at the position of the missing methyl group but presumably does not have an effect either on the stability of the protein or on the enzymatic activity. A comparison of the residues coordinating the two Mg2+ ions shows essentially no difference (the r.m.s. deviation is 0.11 Å for 55 atoms), which suggests that the active site is not altered between the two proteins. Likewise, those 14 residues that were identified in human ARH3 as being likely to participate in the binding of ADP-ribose (Glu25, Asp61, Gly103, Val104, Thr106, Asn126, Phe127, Ser132, Tyr133, Asn135, His166, Glu259, Asp298 and Thr301) have an r.m.s. deviation to the mouse orthologue of only 0.55 Å (for 115 atoms), implying that the binding mode is essentially the same in those two proteins.
Figure 5.

Sequence alignment of ARHs. All three ARH families from M. musculus (mm) and Homo sapiens (hs) were aligned using the program ClustalW (Chenna et al., 2003 ▶); the numbering of residues is according to the mARH3 sequence in the crystal structure (2qty), i.e. without the mitochondrial targeting sequence. Identical residues are shown in red and similar residues are shown in blue. Residues that participate in the coordination of the two Mg2+ ions are underlined. Sequence identities within the family from M. musculus are 16.2% for mARH3 and mARH2, 17.4% for mARH3 and mARH1 and 42.1% for mARH1 and mARH2, while the sequence identity between mARH3 from M. musculus and H. sapiens is 89.7%.
Table 4. Amino-acid substitutions between mouse and human ARH3.
Conservative substitutions are marked with an asterisk after the amino-acid position.
| Amino-acid position | Amino acid in mARH3 | Amino acid in hARH3 | Comment |
|---|---|---|---|
| 4* | Ile | Leu | |
| 22* | Ala | Ser | 6.6 Å from Mg |
| 23* | Val | Phe | |
| 31 | Ser | Asp | |
| 33* | Ala | Thr | |
| 37 | Ser | Arg | |
| 40* | GluA | Gln | Salt bridge to ArgB117 (sym) |
| 52 | Ala | Glu | |
| 56* | Thr | Ala | |
| 66* | Thr | Ala | Located in protein core |
| 105* | Ile | Val | |
| 120* | TyrB | Phe | Hydrogen bond to GluB81 |
| 189 | Val | Glu | |
| 197 | Glu | Lys | |
| 205* | Glu | Asp | |
| 217* | Lys | Arg | |
| 233* | Val | Ile | Located in protein core |
| 240 | Asp | Ala | |
| 241 | Val | Ser | |
| 243* | SerA | Thr | Hydrogen bond to GluA245 |
| SerB | Thr | Hydrogen bond to GluB246 | |
| 275 | His | Asp | |
| 281* | Thr | Ala | |
| 317* | Glu | Asp | |
| 330* | Phe | Tyr | |
| 335* | Val | Ile | |
| 346 | GluB | Lys | Hydrogen bond to HisB341 |
3.4. Comparison of the structural fold of mARH3 to prokaryotic homologues
A protein structure comparison of mARH3 with proteins deposited in the PDB using DALI (Holm & Sander, 1996 ▶) and SSM (Krissinel & Henrick, 2004 ▶) gave the structure of the human orthologue with the highest Z score for both search algorithms (see §3.3 and Table 5 ▶). In addition, two prokaryotic proteins (PDB codes 1t5j from the archaeon Methanococcus janaschii and 2cwc from the Gram-negative eubacterium Thermus thermophilus), as well as two metal complexes of 2cwc (2yzv and 2yzw), were identified using the program SSM with Z scores of 9.4, 8.5 and 8.4, respectively. Like mARH3, they are all-α-helical proteins and show a high degree of structural similarity (82% and 79% of all amino-acid residues can be superimposed with an r.m.s. deviation of 1.4 Å for 246 Cα positions for 1t5j and 240 Cα positions for 2cwc; Table 5 ▶). The proposed active-site crevice shows a high degree of agreement of the positions of the α-helices and loop structures that form this crevice. However, there are also two notable differences in terms of the apparent accessibility to this crevice. An additional α-helix (helix 2) that is present in mouse ARH3 (as well as the human protein) but not in the prokaryotic proteins seems to block access to the active site from this side. The other difference concerns the ‘missing loop’ region (Asp45–Thr54) in mouse ARH3 (and its human orthologue). The corresponding region in 1t5j and 2cwc folds back and forms a kind of lid that blocks access from this side of the active site. To gain full accessibility to the two Mg2+ ions, this lid would need to move away. In the case of 1t5j at least, there is one residue within this lid (Tyr49) that participates in a hydrogen-bonding network together with other Mg2+-coordinating residues.
Table 5. Structural comparison of mARH3 using the search algorithms from DALI (Holm & Sander, 1996 ▶) and SSM (Krissinel & Henrick, 2004 ▶).
Only results with a Z score higher than 2 are listed. The superposition of mARH3 with the corresponding protein was performed with the brute-force method implemented in the program LSQMAN (Novotny et al., 2004 ▶), from which the number of superimposed amino-acid residues, r.m.s. deviation and r.m.s. ΔB factor values were taken.
| Protein name | Organism | PDB code | Annotated function | Space group | Resolution (Å) | No. of amino-acid residues | No. of superimposed Cα positions | Z score | R.m.s. deviation (Å) | R.m.s. ΔB factor (Å2) | Reference† |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ADP-ribosylhydrolase 3 | H. sapiens | 2g4k | De-ADP-ribosylation | P212121 | 1.82 | 336 | 336 | 63.3‡, 15.7§ | 0.47 | 14.5 | a |
| ADP-ribosylhydrolase 3 | H. sapiens | 2fp0 | De-ADP-ribosylation | P21 | 2.05 | 335 | 335 | 15.9§ | 0.58 | 30.0 | b |
| ADP-ribosylhydrolase 3 | H. sapiens | 2foz | De-ADP-ribosylation | P212121 | 1.60 | 336 | 336 | 15.9§ | 0.40 | 6.2 | b |
| Ribosylglycohydrolase MJ1187 | Methanococcus jannaschii | 1t5j | Unknown | P43212 | 2.70 | 301 | 246 | 9.4§ | 1.36 | 34.1 | c |
| ADP-ribosylglycohydrolase | T. thermophilus HB8 | 2cwc | De-ADP-ribosylation | P212121 | 1.65 | 303 | 240 | 8.5§ | 1.44 | 6.5 | d |
| ADP-ribosylglycohydrolase | T. thermophilus HB8 | 2yzw | De-ADP-ribosylation | P212121 | 1.70 | 303 | 241 | 8.5§ | 1.30 | 6.8 | e |
| ADP-ribosylglycohydrolase | T. thermophilus HB8 | 2yzv | De-ADP-ribosylation | P212121 | 1.60 | 303 | 241 | 8.4§ | 1.42 | 6.4 | e |
| Methyl-coenzyme M reductase | Methanobacterium thermoautotrophicum | 1mro | Methanogenesis | P21 | 1.16 | 548 | 69 | 4.2‡ | 2.14 | 4.4 | f |
| Hat domain of murine CstF-77 | Mus musculus | 2ooe | Polyadenylation of mRNA precursors | H32 | 3.00 | 400 | 54 | 2.5‡ | 2.22 | 40.3 | g |
The data for 2cwc and its two metal-ion-containing analogues 2yzv and 2yzw were all collected by the RIKEN structural genomics/proteomics initiative at SPring-8 in Japan at a wavelength of 1.0 Å to rather high resolutions of 1.65 Å (2cwc), 1.60 Å (2yzv) and 1.70 Å (2yzw). The structures of 2yzv, which contains two Mg2+ ions, and of 2yzw, which contains two Gd3+ ions, superimpose very well on each other with an r.m.s. deviation of 0.16 Å for 283 superimposed Cα atoms, as well as with 2cwc with an r.m.s. deviations of 0.24 Å for 284 superimposed Cα atoms for 2yzv and 0.26 Å for 282 superimposed Cα atoms for 2yzw. The two Mg2+ ions within the 2yzv structure are 3.5 Å apart from each other. The first Mg2+ ion, with a B factor of 18 Å2, is coordinated by one Thr and two Asp residues (both coordinating bidentally), while the second ion has a higher B factor of 30 Å2 and is coordinated by one Thr and three Asp residues (one coordinating bidentally). Both ions are not octahedrally coordinated because there are no coordinating water molecules. A comparison with the 2cwc structure shows little structural difference for the three coordinating residues for the first Mg2+ ion, while for the second Mg2+ ion two of the three coordinating Asp residues have shifted by 3.4 and 6.3 Å. Compared with mouse ARH3, there is hardly any difference in the coordination of the first Mg2+ ion, although Glu25 in the ARH3 structure is within coordination distance of this ion. The second Mg2+ ion also shows good agreement compared with ARH3, with the exception that one Asp residue has moved by 1.9 Å.
The situation is similar for 2yzw, which contains two Gd3+ ions 3.76 Å apart from each other with comparable B factors of 13 and 16 Å2. Although the maximal resolution of this structure is somewhat lower than that of 2yzv, both ions are roughly octahedrally coordinated. The first Gd3+ ion is coordinated by two water molecules (of which one is a bridging water molecule between the two metal ions), one Thr, one Glu and two Asp residues (one Asp residue coordinates to both metal ions) and the second Gd3+ ion is coordinated by one Thr and three Asp residues as well as two water molecules. A comparison with the structure of 2cwc shows a large movement of the Glu residue (by 1.8 Å) for the first Gd3+ ion and also a large movement of two of the three coordinating Asp residues. Compared with the mouse ARH3 structure, there is hardly any movement within the coordinating residues of both metal ions apart from a 0.9 Å movement of the coordinating Glu residue.
Using the search algorithm from the program DALI a (weak) structural similarity (Z scores of 4.2 and 2.4, respectively) was detected to two bacterial enzymes (PDB codes 1mro and 2ooe), one of which is involved in bacterial methanogenesis and the other in polyadenylation of mRNA precursors. Given the fact that only 68 out of 548 amino-acid residues can be superimposed with an r.m.s. deviation of 2.06 Å for 1mro (Ermler et al., 1997 ▶) and 54 out of 400 amino-acid residues with an r.m.s. deviation of 2.22 Å for 2ooe (Bai et al., 2007 ▶), it is clear that these proteins do not share a common fold with mARH3.
4. Summary and conclusion
We have determined the three-dimensional crystal structure of murine ADP-ribosylhydrolase 3 to 1.8 Å resolution. The structure constitutes a compact all-α-helical protein with two Mg2+ ions that pinpoint the active-site crevice. A comparison with the recently published structure of the human orthologue (Mueller-Dieckmann et al., 2006 ▶) shows a high degree of structural consistency; similar observations were made using the three-dimensional structures of four prokaryotic proteins deposited in the PDB.
Supplementary Material
PDB reference: ADP-ribosylhydrolase 3, 2qty, r2qtysf
Enhanced figure: interactive version of Fig. 4
Acknowledgments
Parts of the work described in this study represent partial fulfilment of the requirements for the doctoral thesis of SK at the University of Hamburg. This work was supported by grants from the Deutsche Forschungsgemeinschaft (to FK-N and MSW) and the Studienstiftung des deutschen Volkes (to SK).
References
- Ahuja, N., Schwer, B., Carobbio, S., Waltregny, D., North, B. J., Castronovo, V., Maechler, P. & Verdin, E. (2007). J. Biol. Chem.282, 33583–33592. [DOI] [PubMed]
- Aktories, K. & Just, I. (2000). Bacterial Protein Toxins. Berlin: Springer-Verlag.
- Bai, Y., Auperin, T. C., Chou, C.-Y., Chang, G.-G., Manley, J. L. & Tong, L. (2007). Mol. Cell, 25, 863–875. [DOI] [PubMed]
- Chang, P., Jacobson, M. K. & Mitchison, T. J. (2004). Nature (London), 432, 645–649. [DOI] [PubMed]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G. & Thompson, J. D. (2003). Nucleic Acids Res.31, 3497–3500. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Ermler, U., Grabarse, W., Shima, S., Goubeaud, M. & Thauer, R. K. (1997). Science, 278, 1457–1462. [DOI] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Glowacki, G., Braren, R., Firner, K., Nissen, M., Kuehl, M., Reche, P., Bazan, F., Cetkovic Cvrlje, M., Leiter, E., Haag, F. & Koch-Nolte, F. (2002). Protein Sci.11, 1657–1670. [DOI] [PMC free article] [PubMed]
- Haigis, M. C., Mostoslavsky, R., Haigis, K. M., Fahie, K., Christodoulou, D. C., Murphy, A. J., Valenzuela, D. M., Yancopoulos, G. D., Karow, M., Blander, G., Wolberger, C., Prolla, T. A., Weindruch, R., Alt, F. W. & Guarente, L. (2006). Cell, 126, 941–954. [DOI] [PubMed]
- Harding, M. M. (2001). Acta Cryst. D57, 401–411. [DOI] [PubMed]
- Harding, M. M. (2006). Acta Cryst. D62, 678–682. [DOI] [PubMed]
- Hassa, P. O., Haenni, S. S., Elser, M. & Hottiger, M. O. (2006). Microbiol. Mol. Biol.70, 789–829. [DOI] [PMC free article] [PubMed]
- Holm, L. & Sander, C. (1996). Science, 273, 595–603. [DOI] [PubMed]
- Jagtap, P. & Szabo, C. (2005). Nature Rev. Drug Discov.4, 421–440. [DOI] [PubMed]
- Kernstock, S., Koch-Nolte, F., Mueller-Dieckmann, J., Weiss, M. S. & Mueller-Dieckmann, C. (2006). Acta Cryst. F62, 224–227. [DOI] [PMC free article] [PubMed]
- Kim, M. Y., Mauro, S., Gevrey, N., Lis, J. T. & Kraus, W. L. (2004). Cell, 119, 803–814. [DOI] [PubMed]
- Koch-Nolte, F. & Haag, F. (1997). Adv. Exp. Med. Biol.419, 1–13. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Lennon, G. G., Auffray, C., Polymeropoulos, M. & Soares, M. B. (1996). Genomics, 33, 151–152. [DOI] [PubMed]
- Malanga, M. & Althaus, F. R. (2005). Biochem. Cell. Biol.83, 354–364. [DOI] [PubMed]
- Meyer, R. G., Meyer-Ficca, M. L., Whatcott, C. J., Jacobson, E. L. & Jacobson, M. K. (2007). Exp. Cell Res.313, 2920–2936. [DOI] [PMC free article] [PubMed]
- Morrison, A. R., Moss, J., Stevens, L. A., Farrell, C., Merithew, E., Lambright, D. G., Greiner, D. L., Mordes, J. P., Rossini, A. A. & Bortell, R. (2006). J. Biol. Chem.281, 33363–33372. [DOI] [PubMed]
- Moss, J., Jacobson, M. K. & Stanley, S. J. (1985). Proc. Natl Acad. Sci. USA, 82, 5603–5607. [DOI] [PMC free article] [PubMed]
- Moss, J., Zolkiewska, A. & Okazaki, I. (1997). Adv. Exp. Med. Biol.419, 25–33. [DOI] [PubMed]
- Mueller-Dieckmann, C., Kernstock, S., Lisurek, M., von Kries, J.-P., Haag, F., Weiss, M. S. & Koch-Nolte, F. (2006). Proc. Natl Acad. Sci. USA, 103, 15026–15031. [DOI] [PMC free article] [PubMed]
- Mueller-Dieckmann, C., Panjikar, S., Schmidt, A., Mueller, S., Kuper, J., Geerlof, A., Wilmanns, M., Singh, R. K., Tucker, P. A. & Weiss, M. S. (2007). Acta Cryst. D63, 366–380. [DOI] [PubMed]
- Mueller-Dieckmann, J. (2006). Acta Cryst. D62, 1446–1452. [DOI] [PubMed]
- Niere, M., Kernstock, S., Koch-Nolte, F. & Ziegler, M. (2008). Mol. Cell. Biol.28, 814–824. [DOI] [PMC free article] [PubMed]
- Novotny, M., Madsen, D. & Kleywegt, G. J. (2004). Proteins, 54, 260–270. [DOI] [PubMed]
- Oka, S., Kato, J. & Moss, J. (2006). J. Biol. Chem.281, 705–713. [DOI] [PubMed]
- Ono, T., Kasamatsu, A., Oka, S. & Moss, J. (2006). Proc. Natl Acad. Sci. USA, 103, 16687–16691. [DOI] [PMC free article] [PubMed]
- Otto, H., Reche, P. A., Bazan, F., Dittmar, K., Haag, F. & Koch-Nolte, F. (2005). BMC Genomics, 6, 139–161. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Schreiber, V., Dantzer, F., Ame, J. C. & de Murcia, G. (2006). Nature Rev. Mol. Cell Biol.7, 517–528. [DOI] [PubMed]
- Seman, M., Adriouch, S., Haag, F. & Koch-Nolte, F. (2004). Curr. Med. Chem.11, 857–872. [DOI] [PubMed]
- Shall, S. (1995). Biochimie, 77, 313–318. [DOI] [PubMed]
- Smith, S., Giriat, I., Schmitt, A. & de Lange, T. (1998). Science, 282, 1484–1487. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed]
- Yu, W., Ginjala, V., Pant, V., Chernukhin, I., Whitehead, J., Docquier, F., Farrar, D., Tavoosidana, G., Mukhopadhyay, R., Kanduri, C., Oshimura, M., Feinberg, A. P., Lobanenkov, V., Klenova, E. & Ohlsson, R. (2004). Nature Genet.36, 1105–1110. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ADP-ribosylhydrolase 3, 2qty, r2qtysf
Enhanced figure: interactive version of Fig. 4