

The enzyme aspartate semialdehyde dehydrogenase from M. tuberculosis has been expressed, purified and crystallized in two different crystal forms.
Keywords: aspartate semialdehyde dehydrogenase, Mycobacterium tuberculosis, Rv3708c
Abstract
Aspartate semialdehyde dehydrogenase from Mycobacterium tuberculosis (Asd, ASADH, Rv3708c), which is the second enzyme in the lysine/homoserine-biosynthetic pathways, has been expressed heterologously in Escherichia coli. The enzyme was purified using affinity and gel-filtration chromatographic techniques and crystallized in two different crystal forms. Preliminary diffraction data analysis suggested the presence of up to four monomers in the asymmetric unit of the orthorhombic crystal form A and of one or two monomers in the cubic crystal form B.
1. Introduction
The bacterium Mycobacterium tuberculosis is the causative agent of the highly contagious disease tuberculosis (TB). Less than a century ago, TB was still a serious threat to human health even in developed societies, but after the introduction of the BCG vaccine and of the antibiotics isoniazid, rifampin, ethambutol and streptomycin in the 1950s and 1960s the disease was believed to be more or less under control. However, the last two decades have seen a serious re-emergence of TB, especially in patients who are immune-compromised. As a consequence, in 1993 the World Health Organization declared TB a global health emergency. TB is currently responsible for approximately 1.6 million deaths per year (http://www.tballiance.org) and every second someone in the world becomes newly infected with M. tuberculosis. The re-emergence of TB has been mainly attributed to (i) the inefficacy of the BCG vaccine and (ii) the development of drug resistance in M. tuberculosis to virtually all known anti-TB drugs (Shafiani et al., 2005 ▶; World Health Organization, 2007 ▶). Therefore, highly effective new anti-TB drugs need to be developed in order to cope with the problem.
The lysine/homoserine-biosynthetic pathway starts with the amino acid aspartic acid, which is converted to aspartyl-β-phosphate (ABP) by the enzyme aspartokinase. ABP is then further reduced to aspartate-β-semialdehyde (ASA) by the enzyme aspartate-β-semialdehyde dehydrogenase (Asd, ASADH). ASA then either enters into the lysine-biosynthetic pathway to form diaminopimelate (DAP) and lysine or into the homoserine-biosynthetic pathway to form the amino acids isoleucine, methionine and threonine. Thus, the enzyme ASADH plays a major role in both pathways. The absence of the two pathways in humans and the absolute requirement for DAP in bacteria (Galan et al., 1990 ▶; Pavelka & Jacobs, 1996 ▶; Harb & Kwaik, 1998 ▶) and the importance of these four amino acids for protein synthesis make the enzymes in these pathways attractive targets for antibacterial drug discovery (Hutton et al., 2003 ▶).
Studies on Asd from Salmonella typhimurium (Galan et al., 1990 ▶) and Legionella pneumophila (Harb & Kwaik, 1998 ▶) have demonstrated that perturbations of the asd gene encoding Asd are lethal to microbes. Mycobacteria have also been suggested to possess an absolute requirement for this pathway even when growing in rich medium containing DAP and all the metabolic components of the aspartate family (Pavelka & Jacobs, 1996 ▶). Therefore, developing an inhibitor that selectively targets the Asd enzyme appears to be a promising rational approach to controlling the growth of M. tuberculosis (Moore et al., 2002 ▶; Shafiani et al., 2005 ▶).
Structural studies on Asd have been carried out on the enzymes from Escherichia coli [PDB codes 1t4b (Nichols et al., 2004 ▶), 1gl3 (Hadfield et al., 2001 ▶) and 1brm (Hadfield et al., 1999 ▶)], Haemophilus influenza (PDB codes 1nwc and 1nx6; Blanco, Moore, Kalabeeswaran et al., 2003 ▶), Vibrio cholerae (PDB codes 1mb4 and 1mc4; Blanco, Moore & Viola, 2003 ▶), Streptococcus pneumoniae (PDB codes 2gz1, 2gz2, 2gz3 and 2gyy; Faehnle et al., 2006 ▶), Methanococcus jannaschii (PDB code 1ys4, Faehnle et al., 2005 ▶), Thermus thermophilus HB8 (PDB code 2yv3; W. Kagawa, N. Fujikawa, H. Kurumizaka, Y. Bessho, M. J. Ellis, S. V. Antonyuk, R. W. Strange, S. S. Hasnain, S. Kuramitsu & S. Yokoyama, unpublished work) and Pseudomonas aeruginosa (PDB code 2hjs; M. E. Cuff, E. Evdokimova, M. Kudritska, A. Edwards, A. Savchenko & A. Joachimiak, unpublished work) and on the hypothetical Asd from Sulfolobus tokodaii (PDB code 2ep5; Y. Asada & N. Kunishima, unpublished work). In all cases, the oligomeric state of the functional enzyme was observed to be a homodimer. The structure of the Asd monomer is comprised of an N-terminal nucleotide-binding domain and a dimerization domain. The architecture of the N-terminal domain comprises an approximate Rossmann fold. The first β-strand β1 of the N-terminal domain leads into the glycine-rich loop, which is characteristic of an NADP-binding domain. The C-terminal domain is responsible for the dimerization of the enzyme and the binding of the substrate and contains the catalytic residues (Hadfield et al., 1999 ▶, 2001 ▶). The mechanism of enzyme action appears to be the same in all microbes. It proceeds via the attack of the thiol group of the active-site cysteine residue onto the γ-carbon of the substrate l-aspartyl-β-phosphate, producing the acyl-enzyme intermediate and releasing inorganic phosphate. An amino-acid sequence alignment reveals that Asd from M. tuberculosis (Mtb-Asd) is quite different from the Asds from other bacterial pathogens (Shafiani et al., 2005 ▶; Shafiani, 2006 ▶) such as E. coli, S. typhimurium, V. cholerae, H. influenzae and P. aeruginosa. The maximum sequence identities of M. tuberculosis Asd to Asds for which structures have been determined are 42% (Asd from T. thermophilus), 36% (Asd from S. pneumoniae) and 31% (Asd from P. aeruginosa). These values suggest that while the overall structure of Mtb-Asd is likely to be the same as the structure of Asd from any of the other bacterial sources, significant differences are likely to exist in various parts of the enzyme. Here, we report the crystallization of native Mtb-Asd in two different crystal forms and a preliminary X-ray diffraction investigation of these crystal forms.
2. Experimental methods
2.1. Expression and purification of recombinant Asd
The recombinant E. coli M15 strain (Shafiani et al., 2005 ▶) carrying the Mtb asd gene in the expression vector pQE30 (pSST1) was grown in Luria–Bertani (LB) medium supplemented with ampicillin (100 µg ml−1) and kanamycin (25 µg ml−1) under standardized incubation conditions (310 K, 200 rev min−1, overnight). This culture was inoculated to a final concentration of 1%(v/v) in fresh LB medium supplemented with both antibiotics at the same concentrations and incubated at 310 K at 200 rev min−1 for 90 min. The incubation temperature was then decreased stepwise to 303, 298, 293 and 288 K at 90 min intervals. Finally, the culture was incubated at 288 K until an OD600 of about 0.8–1.0 was achieved. At this stage, 0.05 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to induce production of Asd. The induced culture was further incubated (288 K, 18 h, 200 rev min−1) and the cells were then harvested and stored at 193 K until further processing. 1 g of cell pellet was suspended in 10 ml buffer A (20 mM Tris pH 8.0, 150 mM NaCl, 10 mM imidazole, 1 mM DTT, Roche cocktail of protease inhibitors). The cells were lysed by sonication (three cycles, 5 min per cycle, 0.3 s pulses, 277 K). The cell debris was pelleted by centrifugation (20 000g, 60 min, 277 K) and the cell lysate was retained. This lysate was filtered through a 0.45 µm membrane and then passed through a nickel–nitrilotriacetic acid (Ni–NTA) metal-affinity column equilibrated with buffer B (20 mM Tris pH 8.0, 150 mM NaCl, 1 mM DTT). In order to remove unbound proteins, the column was washed with five column volumes of buffer A, with five column volumes of buffer C (20 mM Tris pH 8.0, 1 M NaCl, 10 mM imidazole, 1 mM DTT) and finally with five column volumes of buffer D (20 mM Tris pH 8.0, 150 mM NaCl, 25 mM imidazole). The recombinant Asd protein was eluted using buffer E (20 mM Tris pH 8.0, 150 mM NaCl, 200 mM imidazole, 1 mM DTT) and subsequently purified by gel filtration (Superdex 200, 16/60, Amersham Pharmacia Biotech, calibrated using the Bio-Rad gel-filtration standard) using buffer B for both equilibration and elution. The protein eluted with an apparent molecular weight of approximately 75 kDa, which corresponds to the molecular weight of a homodimer. The peak fractions were analyzed by SDS–PAGE, pooled, concentrated and dialyzed against 20 mM Tris pH 8.0 and 10 mM DTT. The purity of the protein was then gauged by SDS–PAGE. In addition, Asd was also purified in 10 mM potassium phosphate buffer pH 8.0. The purification procedure was identical to that described above except that in all steps the Tris buffer was replaced by phosphate buffer pH 8.0. All other buffer constituents remained the same. The enzymatic activity of purified Asd was checked in a photometric assay according to the procedure described by Shafiani et al. (2005 ▶).
2.2. Crystallization
Purified Asd (concentration 9 mg ml−1) in a buffer composed of 20 mM Tris buffer pH 8.0 and 10 mM DTT was initially screened against Hampton Research Crystal Screens I and II using the hanging-drop vapour-diffusion method. Drops consisting of 2 µl protein solution and 2 µl reservoir solution were equilibrated over 500 µl reservoirs. Plate-like crystals stacked on each other appeared after 1 d at room temperature and within 2 d at 292 K. To obtain single well shaped crystals, dioxane was added in order to slow nucleation. Single crystals were finally obtained using the reservoir composition 4 M sodium formate and 4%(v/v) dioxane (crystal form A; Fig. 1 ▶ a). Purified Asd (concentration 9 mg ml−1) in a buffer composed of 10 mM potassium phosphate buffer pH 8.0 and 10 mM DTT was screened for crystallization using the sitting-drop vapour-diffusion method in the high-throughput crystallization facility operated by EMBL Hamburg, Germany (Mueller-Dieckmann, 2006 ▶). Initial screening experiments were performed against Crystal Screen Cryo, Crystal Screens I and II, Grid Screens PEG 6K, MPD, Ammonium Sulfate and Sodium Malonate, Index Screen, PEG/Ion Screen and Salt Rx Screen from Hampton Research, Basic Screen from Jena Bioscience and the NX_Classic, NX_Classic_Lite, NX_ComPasSuite and NX_Cryo screens from Nextal. The sitting drops consisted of 0.5 µl protein solution and 0.5 µl reservoir solution. They were equilibrated against 80 µl reservoir solution. Very small cube-shaped crystals (crystal form B, Fig. 1 ▶ b) were obtained after 2 d at 292 K using the reservoir composition 1.6 M ammonium sulfate and 100 mM citric acid pH 5.0.
Figure 1.

The two crystal forms of M. tuberculosis Asd. (a) Crystal form A; (b) crystal form B.
2.3. Diffraction data collection and processing
Prior to data collection, a crystal of form A (150 × 120 × 10 µm) was treated with cryoprotectant [20%(v/v) glycerol, 4 M sodium formate] for 30 s and flash-cooled to 100 K. Diffraction data were then collected at EMBL beamline X13 (Hamburg, Germany) using a MAR CCD (165 mm) detector. A crystal of form B (25 × 25 × 25 µm) was treated with cryoprotectant [27%(v/v) glycerol in reservoir buffer] for 30 s and flash-cooled to 100 K. Diffraction data were then collected on ESRF beamline BM14 (Grenoble, France) using a MAR Mosaic (225 mm) CCD detector. The data were indexed and integrated using DENZO (Otwinowski & Minor, 1997 ▶) and scaled using SCALEPACK (Otwinowski & Minor, 1997 ▶). The redundancy-independent merging R factor R r.i.m. as well as the precision-indicating merging R factor R p.i.m. (Weiss, 2001 ▶) were calculated using the program RMERGE (available from http://www.embl-hamburg.de/~msweiss/projects/msw_qual.html or from MSW upon request). The relevant data-collection and processing parameters are given in Table 1 ▶. Intensities were converted to structure-factor amplitudes using the program TRUNCATE (French & Wilson, 1978 ▶; Collaborative Computational Project, Number 4, 1994 ▶). The optical resolution was calculated using the program SFCHECK (Vaguine et al., 1999 ▶; Collaborative Computational Project, Number 4, 1994 ▶).
Table 1. Data-collection and processing statistics.
| Crystal form A | Crystal form B | |
|---|---|---|
| No. of crystals | 1 | 1 |
| Wavelength (Å) | 0.8076 | 1.067 |
| Crystal-to-detector distance (mm) | 200 | 208 |
| Rotation range per image (°) | 0.5 | 0.5 |
| Total rotation range (°) | 204.5 | 36.0 |
| Resolution range (Å) | 99.0–2.70 (2.75–2.70) | 99.0–2.18 (2.22–2.18) |
| Space group | P212121 | F432 |
| Unit-cell parameters (Å, °) | a = 105.69, b = 109.52, c = 175.02 | a = b = c = 267.32 |
| Mosaicity (°) | 0.70† | 0.47 |
| Total no. of reflections | 353558 | 352300 |
| Unique reflections | 55484 | 43078 |
| Redundancy | 6.4 (5.2) | 8.2 (7.5) |
| I/σ(I) | 9.3 (2.5) | 14.1 (2.2) |
| Completeness (%) | 98.2 (90.6) | 99.9 (100.0) |
| Rmerge (%) | 19.0 (80.5) | 15.4 (95.3) |
| Rr.i.m.‡ (%) | 20.6 (88.2) | 16.5 (102.4) |
| Rp.i.m.‡ (%) | 7.9 (35.4) | 5.7 (36.9) |
| Overall B factor from Wilson plot (Å2) | 36.4 | 32.1 |
| Optical resolution§ (Å) | 1.88 | 1.68 |
Unrefined in order to avoid too many overlapping reflections.
Redundancy-independent merging R factor R
r.i.m. = 100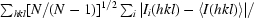
 , with N being the number of times a given reflection hkl was observed; precision-indicating merging R factor R
p.i.m. = 100
, with N being the number of times a given reflection hkl was observed; precision-indicating merging R factor R
p.i.m. = 100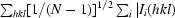 −
− 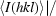 (Weiss, 2001 ▶).
(Weiss, 2001 ▶).
Defined as in Vaguine et al. (1999 ▶).
3. Results and discussion
3.1. Identity, purity and oligomeric state of the protein
The protein construct expressed and purified included the 17-amino-acid sequence RGSHHHHHHGSACELGT between the N-terminal Met and the second amino acid Gly of the native Asd sequence. Thus, the total length of the protein was 362 amino acids and its molecular weight was 38 072 Da. Asd could be purified in either Tris pH 8.0 or phosphate buffer pH 8.0 to a yield of about 10–12 mg protein from the cell pellet obtained from a 1 l bacterial culture. The purity of the sample was approximately 96% as judged by SDS–PAGE (data not shown). Based on the elution profile of the gel filtration, the apparent molecular weight of the protein in solution was 75 kDa (data not shown), which is consistent with the molecular weight of a homodimer. This observation is also consistent with earlier findings on Asds from other organisms, for which the functional unit was established to be a homodimer (Hadfield et al., 1999 ▶; Blanco, Moore, Kalabeeswaran et al., 2003 ▶; Blanco, Moore & Viola, 2003 ▶). The purified Asd was found to be enzymatically active (data not shown).
3.2. Data collection and processing
For crystal form A, a complete X-ray diffraction data set was collected to 2.70 Å resolution (Table 1 ▶). The crystals belong to the orthorhombic space group P212121. All of the approximately 20 crystals screened exhibited high mosaicity, with the mosaicity refining to values larger than 1.5° during post-refinement. Thus, in order to avoid overlapping reflections during data processing, the mosaicity used for integrating the data was set to an artificially low value of 0.7 and not post-refined during the scaling procedure. In this way, it was possible to obtain a nearly complete data set to 2.70 Å resolution without allowing the data quality to degrade too much (Table 1 ▶). Crystals belonging to crystal form B exhibited a significantly lower mosaicity than those of form A. From one form B crystal, a complete and redundant data set was collected to 2.18 Å resolution. The crystal belongs to the cubic space group F432 (Table 1 ▶).
3.3. Analysis of the two crystal forms
Based on the molecular weight of the protein (38 072 Da), the most likely numbers of molecules in the asymmetric unit of the orthorhombic crystal form are four, corresponding to a Matthews parameter V M of 3.33 Å3 Da−1 and a solvent content of 63% (Matthews, 1968 ▶), or six (V M = 2.22 Å3 Da−1, solvent content 45%). Since a self-rotation function displayed only one noncrystallographic twofold axis between the crystallographic y and z axes and since a native Patterson synthesis yielded a non-origin peak at fractional coordinates (u, v, w) = (0.33, 0.46, 0.50) indicating translational symmetry, the most likely number of molecules per asymmetric unit in this crystal form is four. For the cubic crystal form, the most likely number of molecules in the asymmetric unit is two (V M = 2.61 Å3 Da−1, solvent content 53%). However, since neither the self-rotation function nor the native Patterson synthesis displayed any features apart from the expected crystallographic peaks, the presence of just one molecule per asymmetric unit cannot be ruled out with confidence, although this would correspond to a rather unlikely V M of 5.23 Å3 Da−1 and an unusually high solvent content of 76%.
In summary, Asd from M. tuberculosis has been crystallized in two different crystal forms, both of which are suitable for structure determination by X-ray crystallography. The determination of the three-dimensional structure of Mtb-Asd in both crystal forms is currently under way. The comparison of the structure of Mtb-Asd with other Asd structures will certainly shed more light on the molecular mechanism of this important enzyme.
Acknowledgments
We would like to thank Mr Sagar Nimsadkar (Institute of Microbial Technology, Chandigarh, India) for his involvement in the early stages of the project and the DAAD academic exchange programme for providing the financial support for the stay of Mr Rajan Vyas in the laboratory of Dr Manfred S. Weiss at the EMBL Hamburg Outstation, Germany (Grant No. A/07/92519). Also, we would like to thank the DBT (India) for funding this project at Panjab University (BT/PR 7081/PID/06/312/2006) and the ESRF (Grenoble, France) for the allocation and provision of synchrotron beamtime.
References
- Blanco, J., Moore, R. A., Kabaleeswaran, V. & Viola, R. E. (2003). Protein Sci.12, 27–33. [DOI] [PMC free article] [PubMed]
- Blanco, J., Moore, R. A. & Viola, R. E. (2003). Proc. Natl Acad. Sci. USA, 100, 12613–12617. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Faehnle, C. R., Le Coq, J., Liu, X. & Viola, R. E. (2006). J. Biol. Chem.281, 31031–31040. [DOI] [PubMed]
- Faehnle, C. R., Ohren, J. F. & Viola, R. E. (2005). J. Mol. Biol.353, 1055–1068. [DOI] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Galan, J. E., Narakayama, K. & Curtiss, R. (1990). Gene, 94, 29–35. [DOI] [PubMed]
- Harb, O. S. & Kwaik, Y. A. (1998). Infect. Immun.66, 1898–1903. [DOI] [PMC free article] [PubMed]
- Hadfield, A. T., Kryger, G., Ouyang, J., Petsko, G. A., Ringe, D. & Viola, R. E. (1999). J. Mol. Biol.289, 991–1002. [DOI] [PubMed]
- Hadfield, A. T., Shammas, C., Kryger, G., Ringe, D., Petsko, G. A., Ouyang, J. & Viola, R. E. (2001). Biochemistry, 40, 14475–14483. [DOI] [PubMed]
- Hutton, C. A., Southwood, T. J. & Turner, J. J. (2003). Mini Rev. Med. Chem.3, 115–127. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Moore, R. A., Bocik, W. E. & Viola, R. E. (2002). Protein. Expr. Purif.25, 189–194. [DOI] [PubMed]
- Mueller-Dieckmann, J. (2006). Acta Cryst. D62, 1446–1452. [DOI] [PubMed]
- Nichols, C. E., Dhaliwal, B., Lockyer, M., Hawkins, A. R. & Stammers, D. K. (2004). J. Mol. Biol.341, 797–806. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pavelka, M. S. Jr & Jacobs, W. R. Jr (1996). J. Bacteriol.178, 6496–6507. [DOI] [PMC free article] [PubMed]
- Shafiani, S. (2006). PhD thesis. Panjab University, Chandigarh, India.
- Shafiani, S., Sharma, P., Vohra, R. M. & Tewari, R. (2005). J. Appl. Microbiol.98, 832–838. [DOI] [PubMed]
- Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999). Acta Cryst. D55, 191–205. [DOI] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst.34, 130–135.
- World Health Organization (2007). WHO Annual Report. Geneva: World Health Organization.