

The crystal structure of the complex formed by human serum albumin and a bacterial albumin-binding domain with the drug compound naproxen and fatty acid decanoate bound to albumin is described.
Keywords: human serum albumin, GA module, bacterial surface proteins, naproxen, albumin binding, fatty acids
Abstract
The previously determined crystal structure of the bacterial albumin-binding GA module in complex with human serum albumin (HSA) suggested the possibility of utilizing the complex in the study of ligand binding to HSA. As a continuation of these studies, the crystal structure of the HSA–GA complex with the drug molecule naproxen and the fatty acid decanoate bound to HSA has been determined to a resolution of 2.5 Å. In terms of drug binding, the structure suggests that the binding of decanoate to the albumin molecule may play a role in making the haemin site in subdomain IB of the albumin molecule available for the binding of naproxen. In addition, structure comparisons with solved structures of HSA and of the HSA–GA complex show that the GA module is capable of binding to different conformations of HSA. The HSA–GA complex therefore emerges as a possible platform for the crystallographic study of specific HSA–drug interactions and of the influence exerted by the presence of fatty acids.
1. Introduction
Many bacterial species express surface protein domains for binding to host serum proteins (Navarre & Schneewind, 1999 ▶). A well studied example is the PAB (peptostreptococcal albumin-binding) protein, which is expressed on the surface of the Gram-positive anaerobic bacterium Finegoldia magna and which contains a domain for binding to human serum albumin (HSA; de Château et al., 1996 ▶; de Château & Björck, 1994 ▶, 1996 ▶). The albumin-binding domain from F. magna is termed the GA (protein G-related albumin-binding) module. It has been suggested that the acquisition of the ability to recruit serum proteins to its surface increases its pathogenicity by camouflaging the bacteria and possibly also by aiding the bacteria in the capture of albumin-bound nutrients (Johansson et al., 2002 ▶).
The crystal structure of the HSA–GA complex has been described previously (Lejon et al., 2004 ▶) and it was suggested that the readily available HSA–GA crystals could be an entry point for the study of HSA–drug interactions. Such studies could in turn prove useful in the context of improving the pharmacokinetic properties of promising drug candidates by structure-aided design (Colmenarejo, 2003 ▶; Colmenarejo et al., 2001 ▶). In order to investigate the feasibility of obtaining crystal structures of HSA–drug complexes in the presence of fatty acids and the GA module, the HSA–GA complex was cocrystallized in the presence of the fatty acid decanoate and the drug molecule naproxen. The structure of the HSA–GA–naproxen complex has been determined to 2.5 Å resolution. The structure of the complex provides further insights into the interaction between the GA module and HSA and also reveals that albumin-bound fatty acids may exert a significant influence on the binding of naproxen to HSA.
2. Materials and methods
2.1. Protein purification, complex formation and crystallization
The ALB8 gene encoding the GA module was obtained as synthetic DNA in a carrier plasmid and was subcloned into a pET28 vector (Novagen) with an N-terminal histidine tag and thrombin protease cleavage site (HHHHSSGLVPRGSHM). The GA module was overexpressed and was then purified using an Ni2+-loaded chelating column and an imidazole gradient using standard procedures as described elsewhere (Cramer et al., 2007 ▶). Owing to the presence of heterogeneous cleavage products after thrombin digestion, the N-terminal tag was left intact for crystallization. Human serum albumin was obtained from Sigma–Aldrich Co. (catalogue No. A6909) and was purified by gel filtration at 281 K as described previously (Lejon et al., 2004 ▶). Complexes of HSA with decanoate were prepared by cycles of dilution as described by Curry et al. (1998 ▶). Solutions of His-tagged GA module (12 mg ml−1 in 50 mM potassium phosphate pH 7.5) and HSA (100 mg ml−1 in 50 mM potassium phosphate pH 7.5) with decanoate were mixed at room temperature to obtain a final protein solution containing approximately equimolar amounts of histidine-tagged GA module and HSA. (S)-Naproxen (Aldrich, catalogue No. 28478-5) was then incubated with the protein solution for 2–4 h at 295 K at a naproxen:HSA ratio of 6:1. The final naproxen:decanoate molar ratio was approximately 4:1. The approximate final concentrations in the protein solution were 100 mg ml−1 HSA, 12 mg ml−1 GA module, 2.5 mM decanoate and 10 mM naproxen in 50 mM potassium phosphate pH 7.5. Crystals were obtained by hanging-drop vapour diffusion and the crystallization drops consisted of 2 µl protein solution and 2 µl of a crystallization solution containing 26–32% PEG 3350, 50 mM potassium phosphate pH 7.5 and 100 mM ammonium acetate. The crystallization reservoirs contained 1 ml crystallization solution. Crystals appeared after 2–5 d incubation at 293 K. The crystals were found to belong to space group C2, with unit-cell parameters a = 190.5, b = 49.5, c = 79.9 Å, β = 93.0°. The crystals were transferred to crystallization solution containing 10 mM (S)-naproxen and 20% glycerol prior to cryocooling in liquid nitrogen. Screens using other fatty acids [dodecanoate (C12) and tetradecanoate (C14)] were also carried out. Of the screened fatty acids, decanoate was found to be the best fatty-acid additive in terms of reproducible crystallization of the HSA–GA complex.
2.2. Crystallographic data collection, structure determination and refinement
Crystallographic data-collection statistics are summarized in Table 1 ▶. Diffraction data were collected at 100 K at ESRF Grenoble beamline ID14-3. Data were indexed using MOSFLM (Leslie, 1992 ▶) and scaling was performed using SCALA (Evans, 2006 ▶) in the CCP4 interface (Collaborative Computational Project, Number 4, 1994 ▶). Molecular replacement was performed using the program MOLREP (Vagin & Teplyakov, 1997 ▶) and the structure of HSA with decanoate (Bhattacharya, Grüne et al., 2000 ▶) was used as an initial search template (PDB code 1e7e). A second search using the GA chain (PDB code 1tf0, chain B) as a search template yielded a contrast of 6.35 (where a value of greater than 2.5 indicates that a solution has been found; Vagin & Teplyakov, 1997 ▶). Simulated annealing in CNS (Brünger et al., 1998 ▶) of the MOLREP solution was perfomed to reduce model bias. Refinement was performed with REFMAC (Murshudov et al., 1997 ▶) using data to a resolution of 2.5 Å and was cycled with rebuilding in O (Jones et al., 1991 ▶) and Coot (Emsley & Cowtan, 2004 ▶). Coordinates and restraints for the ligands [decanoate and (S)-naproxen] were generated using the SKETCHER and LIBCHECK utilities in the CCP4 interface. The ligands were built into positive difference electron-density maps after a few runs of restrained refinement of the HSA–GA complex. TLS (translation, libration and screw-axis rotation displacement) refinement (Winn et al., 2003 ▶) was incorporated intto the later stages of the refinement process. The final model has an R factor of 23.4% and a free R factor of 28.0%. Data-collection and model refinement statistics are summarized in Table 1 ▶. The geometry of the final model was analysed using PROCHECK (Laskowski et al., 1993 ▶). Least-squares superpositions for structure comparisons were performed with LSQMAN (Kleywegt & Read, 1997 ▶).
Table 1. Crystallographic statistics.
Values in parentheses are for the highest resolution shell.
| Unit-cell parameters (Å, °) | a = 190.5, b = 49.5, c = 79.9, β = 93.0 |
| Resolution range (Å) | 94.92–2.5 (2.58–2.5) |
| No. of reflections | 23573 (1294) |
| Multiplicity | 6.3 (3.7) |
| Completeness (%) | 99.0 (98.3) |
| 〈I/σ(I)〉† | 12.2 (2.8) |
| Rmerge‡ (%) | 14.0 (36.1) |
| Wilson B (Å2) | 58.6 |
| Protein residues | 636 |
| Ligand molecules | 7 |
| Rwork/Rfree§ (%) | 23.4/28.0 |
| No. of TLS groups | 6 |
| Average B values (Å2) | |
| Overall | 61.9 |
| Main chain | 60.8 |
| Side chains | 62.9 |
| Ligands | 53.6 |
| R.m.s.d. bond lengths (Å) | 0.017 |
| R.m.s.d. bond angles (°) | 1.371 |
| Ramachandran statistics | |
| Residues in core regions (%) | 89.5 |
| Residues in disallowed regions (%) | 0.2 |
〈I/σ(I)〉 is the average of the intensity divided by its standard deviation.
R
merge = 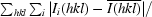
 , where Ii(hkl) is the ith intensity measurement of reflection hkl and
, where Ii(hkl) is the ith intensity measurement of reflection hkl and 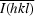 is the weighted average of all measurements of I.
is the weighted average of all measurements of I.
R
work = 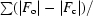
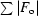 , where F
o and F
c are the observed and calculated structure-factor amplitudes. R
free is the same as R
work but for 1–3% of the data that were omitted from refinement.
, where F
o and F
c are the observed and calculated structure-factor amplitudes. R
free is the same as R
work but for 1–3% of the data that were omitted from refinement.
3. Results and discussion
3.1. Preparation of the HSA–GA complex
The HSA–GA complex described in this study was obtained using a recombinantly expressed GA module that was purified according to standard protocols (Cramer et al., 2007 ▶). Our previous observations of a fatty acid bound to albumin near the GA-binding site in the crystal structure of the HSA–GA complex also prompted the inclusion of the saturated fatty acid decanoate in the preparation of HSA–GA complexes. During crystallization trials, it was found that the addition of decanoate resulted in reproducible crystallization of the HSA–GA complex. Crystals of the complex could not be obtained in the absence of decanoate.
3.2. Structure determination
Initial attempts to solve the structure by molecular replacement using the previously determined structure of the HSA–GA complex (Lejon et al., 2004 ▶) did not yield a satisfactory solution. The structure was instead solved using the albumin chain from the crystal structure of HSA with decanoate as an initial search model and the GA chain from the HSA–GA complex as a second search model.
The use of translation, libration and screw-axis rotation (TLS) displacement parameters to describe the rigid-body motion of the flexible domains of the HSA molecule proved beneficial during the final stages of refinement.
3.3. Overall structure
The overall structure of the HSA–GA–naproxen complex is shown in Fig. 1 ▶. The model comprises HSA residues 6–80 and 86–584 and GA residues 1–53. Two N-terminal histidine-tag residues in the GA chain are also included in the model. After refinement of the HSA and GA coordinates, F o − F c electron-density maps calculated with the phase angles derived from the models of HSA and GA allowed the modelling of ligands bound to HSA. Six molecules of decanoate have been built in fatty-acid sites FA1–4, FA6 and FA7 (notation according to Bhattacharya, Curry et al., 2000 ▶) in the albumin molecule. Fatty acid-binding sites have previously been shown to overlap with drug sites 1 and 2 in HSA (Honoré & Brodersen, 1988 ▶; Bhattacharya, Grüne et al., 2000 ▶). In the HSA–GA–naproxen structure (this work) both drug site 1 (fatty acid-binding site FA7) and drug site 2 (FA3 and FA4) are occupied by fatty acids (Figs. 2 ▶ b and 2 ▶ c).
Figure 1.

Overall structure of the HSA–GA–naproxen complex. Domain II in HSA (residues 197–383) is coloured brown. Bound ligands are shown as spheres, with naproxen in cyan and decanoate in green. The notation of the binding sites is according to Ghuman et al. (2005 ▶).
Figure 2.

Ligand binding in the HSA–GA–naproxen complex. The HSA chain is shown in light orange and the GA module is shown in light blue. Electron density for fatty acids is shown as 2m|F o| − D|F c| OMIT maps contoured at 0.21 e Å−3. (a) The fatty acid in FA6 near the GA-binding site. (b) The fatty acid in drug site 1 (FA7). The orientation of the fatty acid carboxylate moiety is not clear and has not been modelled. (c) The fatty acids in drug site 2 (FA3, FA4). (d) Naproxen binding in the haemin site. Naproxen is shown as cyan sticks, with a 2m|F o| − D|F c| electron-density map contoured at 0.21 e Å−3 (1.0σ) covering the molecule. Residues 104–113 have been removed for clarity.
Difference electron density in domain IB clearly shows the location of the naphthalene ring of naproxen, with density also indicating the orientation of the methoxy and propanoate groups (Fig. 2 ▶ d), allowing unambiguous modelling of one molecule of naproxen in this site.
The final model has 89.5% of its residues in the core region of the Ramachandran plot. There is one residue in the disallowed region of the Ramachandran plot and this residue (Glu95) is located in a region with poor density close to a crystal contact zone. Domain I of the HSA molecule (residues 6–195) is relatively flexible compared with domains II and III. This flexibility is reflected by poorer electron density for this region and several side chains have been modelled with zero occupancy to indicate the uncertainty in their conformation.
3.4. Fatty acid-binding sites
In addition to decanoate binding in drug sites 1 and 2, the electron density clearly indicates the presence of a fatty acid bound to fatty-acid site 6 (FA6) at the interface between subdomains IIA and IIB (Fig. 2 ▶ a). This echoes the observation in the structure of the HSA–GA complex, which also contained fatty acids in FA6. However, whereas the previously reported HSA–GA structure contains two molecules of decanoate modelled in a tail-to-tail arrangement in site FA6, the electron density for the present HSA–GA–naproxen structure indicates that only one molecule of decanoate is bound in this site. As a result, the single fatty acid binds in a slightly altered conformation which is similar to the conformation reported for fatty acids with chain lengths of C14–C18 binding in this site (Bhattacharya, Curry et al., 2000 ▶), but with the carboxylate moiety positioned near the surface of domain II rather than towards the centre. The conformation in the present structure thus allows a direct interaction between the fatty acid and the GA module in which the carboxylate moiety of the decanoate molecule forms a hydrogen bond to the side chain of Glu47. This is in contrast to the previously determined HSA–GA structure, which only suggested an indirect role for the fatty acid in inducing a rotation of the side chain of Lys212 from HSA to form a hydrogen bond to Glu47 from GA (Fig. 3 ▶).
Figure 3.

Close-up of the HSA–GA interface. 187 Cα atoms in domain II of HSA in the structure of the HSA–GA complex (all atoms cyan) and the HSA–GA–naproxen structure (albumin chain in light orange, the GA module in light blue and decanoate in green) have been superposed. Proposed hydrogen bonds are indicated with dashed lines. Interatomic distances for proposed hydrogen bonds are given in angstroms. Electron density for the HSA–GA–naproxen structure is shown as a 2m|F o| − D|F c| OMIT map contoured at 0.21 e Å−3 (1.0σ).
3.5. Naproxen-binding site
Naproxen, or (S)-(6-methoxynaphthalen-2-yl)propanoic acid, is a nonsteroidal anti-inflammatory drug (NSAID) that is widely used to reduce pain, inflammation and fever. Published binding data have indicated that in the absence of fatty acids naproxen binds in drug sites 1 and 2 (Sjöholm et al., 1979 ▶) on HSA, with drug site 2 (Sudlow et al., 1976 ▶) as the highest affinity site. Naproxen has been reported to bind to HSA with an association constant K a of 1.2–1.8 × 106 M −1, leaving <1% of the administered drug free in the plasma (Kober & Sjöholm, 1980 ▶; Honoré & Brodersen, 1984 ▶). Located in subdomain IIIA, drug site 2 involves the hydroxyl group of Tyr411 as the main interaction partner for drug binding. Studies have also reported competitive binding between naproxen and ibuprofen, suggesting that the sites should overlap (Kober & Sjöholm, 1980 ▶). A published crystal structure of ibuprofen complexed with defatted HSA showed that binding of ibuprofen in this site primarily involves interactions with Arg410, Tyr411 and Ser489 (Ghuman et al., 2005 ▶).
The crystal structure of the HSA–GA–naproxen complex reported here shows that drug sites 1 and 2 are occupied by the fatty acid decanoate and that naproxen instead binds in a binding pocket in domain IB in HSA. The binding pocket in domain IB is lined by the side chains of Leu115, Leu135, Ile142, Phe149, Leu154 and Phe157 and the methylene chains of Arg186 and Lys190. This binding site has previously been identified to be the primary binding site for haemin (iron-containing porphyrin) in HSA (Wardell et al., 2002 ▶; Zunszain et al., 2003 ▶). This site has also been suggested to be a secondary binding site for the NSAID compounds azapropazone and indomethacin and the aspirin analogue triiodobenzoic acid (Ghuman et al., 2005 ▶).
In the present structure, one molecule of decanoate is bound in an adjacent compartment of the binding pocket (denoted fatty-acid site FA1), with its carboxylate moiety anchored by a hydrogen bond to Arg117 and its nonpolar tail sandwiched between Tyr138 and Tyr161. In contrast to other drugs that bind in this site, naproxen does not participate in hydrogen bonds with the side chains of albumin residues. Instead, binding seems to be mainly governed by interactions between the aromatic naphthalene and the apolar side chains lining the pocket. The hydrocarbon tail of the neighbouring decanoate is also within interaction distance of the naproxen molecule, interacting in a similar manner as observed for azapropazone, indomethacin and triiodobenzoic acid, which were also bound parallel to a fatty acid in FA1.
With regard to naproxen binding, simultaneous binding of decanoate to HSA seems to exert a significant influence. Neither the 4:1 molar ratio of naproxen to decanoate present in the crystallization solution or the large excess of naproxen added to the crystals immediately prior to cryocooling seem to have been sufficient to displace the decanoate molecules that have bound in the expected binding site for naproxen, i.e. in drug site 2. There are no published reports that the haemin site might be a secondary low-affinity binding site for naproxen, but considering that solution binding studies of naproxen and ibuprofen with HSA were mainly conducted in the absence of fatty acids, there is a possibility that fatty acids could influence the extent to which naproxen is bound to HSA in drug site 2. Previous structural studies of the interactions between HSA and other drugs have confirmed that competition with fatty acids for binding sites may affect the extent and location of drug binding to HSA (Ghuman et al., 2005 ▶; Petitpas et al., 2003 ▶). It is therefore likely that the presence of fatty acids bound to HSA could make other sites available for binding naproxen.
3.6. Structure comparisons
The binding of decanoate and naproxen to the albumin molecule does not seem to interfere with the binding of the GA module to its previously characterized binding site in HSA (Lejon et al., 2004 ▶). The overall assembly of the protein components of the HSA–GA–naproxen complex is therefore highly similar to the crystal structure of the HSA–GA complex, with the GA module forming a three-helix bundle that packs against helices on the exterior of domain II of the albumin molecule (Fig. 1 ▶). Also, a comparison of the current structure of the HSA–GA–naproxen complex with the HSA–GA structure shows no significant change in the conformation of the GA chain.
In contrast, the overall conformation of the albumin chain shows higher structural similarity to a HSA–decanoate structure (PDB code 1e7e) than to the structure of the albumin chain in the HSA–GA complex (PDB code 1tf0). Least-squares superpositioning of 555 Cα atoms from all domains of the HSA chains in the present HSA–GA–naproxen structure onto the HSA–decanoate structure yields a root-mean-square distance (r.m.s.d.) value of 1.57 Å. The corresponding r.m.s.d. value for superposition of the albumin chain in the HSA–GA–naproxen structure onto the albumin chain in the structure of the HSA–GA complex is 3.21 Å.
Based on previous observations of fatty acid-induced conformational changes in HSA (Curry et al., 1998 ▶), it is likely that the difference in global conformation of the albumin chain in the two structures of the HSA–GA complex is a result of the difference in fatty-acid content. The albumin chain in the previously solved HSA–GA structure, which displays greater structural similarity to the structure of defatted albumin than to the structure of albumin with decanoate bound, only shows electron density consistent with a total of three fatty-acid molecules bound to the albumin molecule and no fatty acid bound in FA2. In contrast, the current structural study of the HSA–GA complex shows clear evidence for six molecules of decanoate, including one molecule bound in FA2. The accommodation of a fatty acid in FA2 has previously been suggested to play the chief role in driving the conformational change in HSA upon binding fatty acids (Curry et al., 1998 ▶, 1999 ▶). The global conformational differences in the albumin chains in the two HSA–GA structures are depicted in Fig. 4 ▶(a).
Figure 4.

Structure comparisons. The structures of a HSA–decanoate complex (magenta, PDB code 1e7e), the HSA–GA complex (cyan, PDB code 1tf0) and the current HSA–GA–naproxen structure (in black) are shown in loop representations. 187 Cα atoms from domain II in HSA (residues 197–383) were superposed. The rotations of domains I and III relative to the HSA–GA–naproxen structure are indicated by arrows. (a) Superposition of the HSA–GA complex and the HSA–GA–naproxen complex. (b) Superposition of the HSA–decanoate complex and the HSA–GA–naproxen complex.
The physiological relevance of studying the influence of the medium-chain fatty acid decanoate (C10) on binding conditions for GA in serum may be argued to be less than for long-chain fatty acids (C16–C20) (Peters, 1995 ▶). However, it is worth pointing out that the altered binding mode of the decanoate molecule bound in the HSA–GA–naproxen structure shows that GA binding is not conditional on the exact conformation of the decanoate bound in this site. It is therefore possible that GA binding could be similar even in the presence of a longer chain fatty acid in site FA6. The crystallization trials in this study mainly explored decanoate for HSA–GA complex formation. The fatty acids dodecanoate and tetradecanoate also resulted in crystallization of the HSA–GA complex. Further investigations using long-chain and/or unsaturated fatty acids could shed further light on the influence of the fatty acid at the HSA–GA interface on complex formation.
4. Conclusions
In summary, the structure presented here suggests that the binding of fatty acids to HSA is an important prerequisite for the binding of GA to HSA and also highlights that the fatty-acid content of the albumin molecule might influence the binding mode of drug molecules to HSA in vivo in unpredictable ways. Given that HSA carries 0.1–2 mol of fatty acid per mole of albumin under normal physiological conditions (Fredrickson & Gordon, 1958 ▶; Bhattacharya, Curry et al., 2000 ▶), the continued study of interactions between HSA and GA in the presence of fatty acids or other molecules that bind to HSA may not only shed more light on the conditions for GA binding, but may perhaps also help in the structural studies of HSA–ligand interactions in the presence of fatty acids. The structure opens the possibility of studying the ligand-binding properties of HSA by the crystallization of HSA–GA complexes with varying levels of fatty acids simultaneously bound to albumin. Further studies to investigate the consistency of the binding of drug molecules and long-chain fatty acids to the HSA–GA complex will be valuable in this respect.
The structure also highlights the versatility of the GA module in its interaction with HSA. It would seem that a certain tolerance for the flexibility of HSA is beneficial to the invading bacterial pathogen in this case, enabling it to take advantage of the high concentration (>0.6 mM) of HSA in plasma, regardless of the conformation adopted by the flexible binding partner. As mentioned above, this tolerance for flexibility may also be useful in terms of exploiting the HSA–GA complex as a platform for the study of HSA–ligand interactions.
Supplementary Material
PDB reference: human serum albumin with S-naproxen and the GA module, 2vdb, r2vdbsf
Acknowledgments
We wish to thank Tomas Lundqvist and Niek Dekker at SCL, AstraZeneca, Mölndal, Sweden for help with the cloning and purification of the GA module. We are also grateful to the European Synchrotron Radiation Facility in Grenoble for beam time and assistance, and to Professor Janos Hajdu, Uppsala University for project assistance. This work was supported by grants from the Swedish Research Council to JH.
References
- Bhattacharya, A. A., Curry, S. & Franks, N. P. (2000). J. Biol. Chem.275, 38731–38738. [DOI] [PubMed]
- Bhattacharya, A. A., Grüne, T. & Curry, S. (2000). J. Mol. Biol.303, 721–732. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Château, M. de & Björck, L. (1994). J. Biol. Chem.269, 12147–12151. [PubMed]
- Château, M. de & Björck, L. (1996). Proc. Natl Acad. Sci. USA, 93, 8490–8495. [DOI] [PMC free article] [PubMed]
- Château, M. de, Holst, E. & Björck, L. (1996). J. Biol. Chem.271, 26609–26615. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Colmenarejo, G. (2003). Med. Res. Rev.23, 275–301. [DOI] [PubMed]
- Colmenarejo, G., Alvarez-Pedraglio, A. & Lavandera, J. L. (2001). J. Med. Chem.44, 4370–4378. [DOI] [PubMed]
- Cramer, J. F., Nordberg, P. A., Hajdu, J. & Lejon, S. (2007). FEBS Lett.17, 3178–3182. [DOI] [PubMed]
- Curry, S., Brick, P. & Franks, N. P. (1999). Biochim. Biophys. Acta, 1441, 131–140. [DOI] [PubMed]
- Curry, S., Mandelkow, H., Brick, P. & Franks, N. (1998). Nature Struct. Biol.5, 827–835. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fredrickson, D. S. & Gordon, R. S. (1958). J. Clin. Invest.37, 1504–1515. [DOI] [PMC free article] [PubMed]
- Ghuman, J., Zunszain, P. A., Petitpas, I., Bhattacharya, A. A., Otagiri, M. & Curry, S. (2005). J. Mol. Biol.353, 38–52. [DOI] [PubMed]
- Honoré, B. & Brodersen, R. (1984). Mol. Pharmacol.25, 137–150. [PubMed]
- Honoré, B. & Brodersen, R. (1988). Anal. Biochem.171, 55–66. [DOI] [PubMed]
- Johansson, M. U., Frick, I.-M., Nilsson, H., Kraulis, P. J., Hober, S., Jonasson, P., Linhult, M., Nygren, P.-A., Uhlén, M., Björck, L., Drakenberg, T., Forsén, S. & Wikström, M. (2002). J. Biol. Chem.277, 8114–8120. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kleywegt, G. J. & Read, R. J. (1997). Structure, 5, 1557–1569. [DOI] [PubMed]
- Kober, A. & Sjöholm, I. (1980). Mol. Pharmacol.18, 421–426. [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lejon, S., Frick, I.-M., Björck, L., Wikström, M. & Svensson, S. (2004). J. Biol. Chem.279, 42924–42928. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Navarre, W. W. & Schneewind, O. (1999). Microbiol. Mol. Biol. Rev.63, 174–229. [DOI] [PMC free article] [PubMed]
- Peters, T. (1995). All About Albumin: Biochemistry, Genetics and Medical Applications. San Diego: Academic Press.
- Petitpas, I., Petersen, C. E., Ha, C. E., Bhattacharya, A. A., Zunszain, P. & Ghuman, J. (2003). Proc. Natl Acad. Sci. USA, 100, 6440–6445. [DOI] [PMC free article] [PubMed]
- Sjöholm, I., Ekman, B., Kober, A., Ljungstedt-Påhlman, I., Seiving, B. & Sjödin, T. (1979). Mol. Pharmacol.16, 767–777. [PubMed]
- Sudlow, G., Birkett, D. J. & Wade, D. N. (1976). Mol. Pharmacol.12, 1052–1061. [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Wardell, M., Wang, Z., Ho, J. X., Robert, J., Ruker, F., Ruble, J. & Carter, D. C. (2002). Biochem. Biophys. Res. Commun.291, 813–819. [DOI] [PubMed]
- Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Methods Enzymol.374, 300–321. [DOI] [PubMed]
- Zunszain, P., Ghuman, J., Komatsu, T., Tsuchida, E. & Curry, S. (2003). BMC Struct. Biol.3, 6. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human serum albumin with S-naproxen and the GA module, 2vdb, r2vdbsf