

To investigate the mechanism by which P58(IPK) functions to promote protein folding within the ER, a P58(IPK) TPR fragment without the C-terminal J-domain has been crystallized.
Keywords: P58(IPK), unfolded protein responses, endoplasmic reticulum, stress
Abstract
Endoplasmic reticulum (ER) stress induces the unfolded protein response (UPR), which can promote protein folding and misfolded protein degradation and attenuate protein translation and protein translocation into the ER. P58(IPK) has been proposed to function as a molecular chaperone to maintain protein-folding homeostasis in the ER under normal and stressed conditions. P58(IPK) contains nine TPR motifs and a C-terminal J-domain within its primary sequence. To investigate the mechanism by which P58(IPK) functions to promote protein folding within the ER, a P58(IPK) TPR fragment without the C-terminal J-domain was crystallized. The crystals diffract to 2.5 Å resolution using a synchrotron X-ray source. The crystals belong to space group P21, with unit-cell parameters a = 83.53, b = 92.75, c = 84.32 Å, α = 90.00, β = 119.36, γ = 90.00°. There are two P58(IPK) molecules in the asymmetric unit, which corresponds to a solvent content of approximately 60%. Structure determination by MAD methods is under way.
1. Introduction
Endoplasmic reticulum (ER) stress, which is caused by the threat of protein misfolding within the ER lumen, induces the so-called unfolded protein response (UPR; Ron & Walter, 2007 ▶; Schroder & Kaufman, 2005 ▶). UPR responses are initiated by several ER-resident stress-sensor proteins (IRE1, PERK and ATF-6) through interactions with the ER chaperone BiP (Wu et al., 2007 ▶; Lin et al., 2007 ▶; Marciniak et al., 2006 ▶). The UPR can lower the ER stress burden by regulating a number of transcription pathways. Two major steps are taken by the UPR to deal with ER stress. One is to reduce the ER protein influx and the second is to promote protein folding and the degradation of existing unfolded and misfolded proteins (Kang et al., 2006 ▶; Hollien & Weissman, 2006 ▶; Rutkowski & Kaufman, 2007 ▶; Ron & Walter, 2007 ▶). Eventually, the effects of ER stress may be cleared by the UPR and the homeostasis of the ER re-established. When this fails, the cell may go through apoptosis (Lin et al., 2007 ▶). Malfunctions in UPR pathways may contribute to human diseases such as diabetes and neurodegeneration (Marciniak & Ron, 2006 ▶; Malhotra & Kaufman, 2007 ▶).
P58(IPK), also known as DnaJC3, plays major roles in the UPR during ER stress by carrying out multiple functions. P58(IPK) knockout may cause elevated amounts of misfolded protein within the ER and higher levels of UPR signaling (Oyadomari et al., 2006 ▶). Mutations in P58(IPK) may generate diabetes symptoms in a mouse model (Ladiges et al., 2005 ▶). P58(IPK) has an N-terminal signal sequence for ER targeting and translocation (Rutkowski et al., 2007 ▶). Recent studies have shown that P58(IPK) may function as a molecular chaperone in association with BiP to facilitate the folding of misfolded proteins within the ER (Rutkowski et al., 2007 ▶). The major ER molecular chaperone BiP might be recruited to the scene of protein misfolding by the C-terminal J-domain of P58(IPK). P58(IPK) may also play an important role in co-translational degradation at the ER translocon site for misfolded proteins (Oyadomari et al., 2006 ▶). On the cytosolic side, P58(IPK) was initially identified as an inhibitor of virally induced eIF2α kinase PKR (Barber et al., 1994 ▶). Subsequently, P58(IPK) was also shown to be able to bind and inhibit the ER stress-inducible eIF2α kinase PERK (Yan et al., 2002 ▶; van Huizen et al., 2003 ▶). PKR and PERK function to attenuate protein synthesis by regulating the phosphorylation of translation initiation (Ron & Harding, 2007 ▶). Therefore, P58(IPK) may help to re-establish protein synthesis late in the UPR by inhibiting PKR and PERK.
P58(IPK) contains an N-terminal ER-targeting signal sequence, nine tetratricopeptide repeats (TPRs) and a C-terminal J-domain, as predicted by the primary sequence. The J-domain was first identified within the Hsp40 DnaJ sequence and has become a hallmark of all Hsp40 proteins (Hartl, 1996 ▶; Bukau & Horwich, 1998 ▶). The J-domain contains about 70 amino-acid residues and can interact with Hsp70 to stimulate the Hsp70 ATPase activity. P58(IPK) may recruit Hsp70 and stimulate the Hsp70 ATPase activity for subsequent protein refolding through the C-terminal J-domain (Melville et al., 1999 ▶). The bulk of P58(IPK) contains nine TPR motifs. It has been suggested that P58(IPK) has the potential to function as a molecular chaperone that interacts with misfolded proteins (Oyadomari et al., 2006 ▶). Therefore, the TPR motifs of P58(IPK) may act as the binding site for the misfolded protein. In addition, some TPR motifs have been reported to bind directly to the negatively charged C-terminus of Hsp70 or Hsp90 (Scheufler et al., 2000 ▶; Young et al., 2003 ▶). It is possible that P58(IPK) may utilize its large TPR region to recruit the molecular chaperones Hsp70/Hsp90 to promote the refolding of misfolded proteins.
2. Experimental and discussion
2.1. Cloning, expression and purification of mouse P58(IPK)
The gene encoding the mouse P58(IPK) TPR fragment (residues 33–393) without the N-terminal ER-targeting signal and the C-terminal J-domain was amplified by PCR. The full-length mouse P58(IPK) was utilized as the template. The PCR product was digested using the restriction enzymes NdeI and XhoI. The digested PCR product was then ligated into pET28b by T4 ligase. The P58(IPK) fragment sequence was confirmed by DNA sequencing. The plasmid was then transformed into Escherichia coli strain BL21 (DE3) for protein expression.
The E. coli cells were harvested 20 h after induction with 0.5 mM IPTG at 291 K. Because the recombinant P58(IPK) fragment was histidine-tagged, it could be relatively easily purified using a metal-chelating column. The supernatant was pumped through an Ni-charged column containing about 10 ml resin. The column was thoroughly washed with 50 mM Tris buffer pH 7.9, 0.5 M NaCl and 50 mM imidazole to remove contaminating proteins. The bound protein was then eluted with 50 mM Tris buffer pH 7.9, 0.5 M NaCl and 200 mM imidazole. After Ni-column purification, the N-terminal histidine tag of P58(IPK) was then released by thrombin treatment. The recombinant P58(IPK) was further purified on a Superdex 200 gel-filtration column (GE Healthcare) mounted on an ÄKTA HPLC system (GE Healthcare) to remove thrombin and digested peptides. The apparent molecular weight of the P58(IPK) was shown to be about 40 kDa based on the protein elution time from the gel-filtration column, indicating that the P58(IPK) fragment forms a monomer in solution. The typical yield of purified soluble P58(IPK) fragment from 1 l culture is ∼20 mg (Fig. 1 ▶ a). The mass spectrum of the protein (41 593.6 Da by MALDI) indicated that the protein contained the P58(IPK) TPR fragment (residues 33–393).
Figure 1.

(a) A 13% SDS–PAGE gel of the purified mouse P58(IPK) fragment. The left lane contains protein standards of 97.4, 66, 45, 31, 21.5 and 14.5 kDa (from top to bottom). The right lane contains the purified mouse P58(IPK) fragment. (b) P58(IPK) fragment crystals. (c) Diffraction pattern of a P58(IPK) fragment crystal shown using the HKL-2000 package. The resolution at the detector edge is 2.50 Å.
2.2. Crystallization, data collection and processing
The P58(IPK) fragment protein was concentrated to 20 mg ml−1 in 10 mM Tris buffer pH 8.0, 150 mM NaCl and subjected to crystallization trials. The hanging-drop vapor-diffusion method was used for the crystallization trials. 2 µl protein solution was mixed with 2 µl mother liquor to constitute the hanging drop. Large plate-shaped crystals (0.3 × 0.3 × 0.05 mm) were obtained by the hanging-drop vapor-diffusion method using Linbro plates at room temperature (Fig. 1 ▶ b). The well solution consisted of 1 ml 100 mM HEPES buffer pH 7.0, 15% PEG 5000 MME. The P58(IPK) fragment crystals grew to full size within 2 d.
Diffraction data were collected on SER-CAT beamline 22-ID at the APS. The crystal was flash-frozen at 100 K in a nitrogen-gas stream using a cryoprotectant consisting of 100 mM HEPES buffer pH 7.0, 15% PEG 5000 MME and 20% glycerol. The crystals were soaked in the cryoprotectant for about 30 s before being transferred into the cold stream.
The P58(IPK) fragment crystals diffracted X-rays to 2.5 Å resolution at SER-CAT (Fig. 1 ▶ c). The wavelength was set at 1.0 Å. The data were collected using a MAR300 CCD detector. During data collection, the crystal-to-detector distance was maintained at 380 mm. 200 images covering an oscillation range of 200° were collected and processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). The crystals belong to space group P21, with unit-cell parameters a = 83.53, b = 92.75, c = 84.32 Å, α = 90.00, β = 119.36, γ = 90.00°. The R merge of the data set is 6.0%. The details of the data set are shown in Table 1 ▶. Crystal analysis shows that the asymmetric unit contains two molecules of the P58(IPK) fragment, which corresponds to a solvent content of 64% (V M = 3.41 Å3 Da−1).
Table 1. Statistics of the data set from the P58(IPK) crystals.
| Resolution shell (Å) | I/σ(I) | Rmerge† | Completeness (%) | Redundancy |
|---|---|---|---|---|
| 50.00–4.80 | 29.2 | 0.054 | 83.8 | 6.0 |
| 4.80–3.81 | 27.3 | 0.046 | 88.4 | 5.8 |
| 3.81–3.33 | 22.0 | 0.057 | 89.6 | 5.7 |
| 3.33–3.02 | 16.2 | 0.072 | 91.2 | 5.6 |
| 3.02–2.81 | 10.7 | 0.100 | 91.6 | 5.6 |
| 2.81–2.64 | 6.74 | 0.138 | 92.3 | 5.5 |
| 2.64–2.50 | 4.57 | 0.184 | 89.2 | 5.3 |
| Overall | 20.7 | 0.060 | 89.4 | 5.6 |
R
merge = 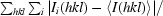
 .
.
A Patterson map search performed using the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) did not reveal any significant peaks other than the origin peak, which indicates the absence of pseudo-translation between the two P58(IPK) molecules within the asymmetric unit. The self-rotation function map did not show any significant peaks in the κ = 180° section.
3. Discussion
P58(IPK) is an important target of the UPR and is transcriptionally activated during ER stress. In association with ER Hsp70 BiP, P58(IPK) may function as a molecular chaperone to promote non-native protein refolding. The mechanisms by which P58(IPK) interacts with misfolded proteins and it putative partner, the molecular chaperone BiP, are currently unknown. The crystal structure of P58(IPK) is critically needed to understand the molecular basis of its functions. In this study, the mouse P58(IPK) TPR fragment was purified and crystallized. The crystals diffracted X-rays to 2.5 Å resolution on the SER-CAT beamline at APS. We propose to determine the P58(IPK) fragment crystal structure using the MAD method. A SeMet P58(IPK) fragment has been produced and we are currently working on the structure determination of the SeMet P58(IPK) TPR fragment.
Acknowledgments
We are grateful to the staff scientists at the SER-CAT beamline at APS for their help during data collection. This work was supported by grants from NIH (R01 DK56203 and R01 GM080261) and the Army Research Office (51894LS) to BS and NIH grants DK47119 and ES08681 to DR.
References
- Barber, G. N., Thompson, S., Lee, T. G., Strom, T., Jagus, R., Darveau, A. & Katze, M. G. (1994). Proc. Natl Acad. Sci. USA, 91, 4278–4282. [DOI] [PMC free article] [PubMed]
- Bukau, B. & Horwich, A. L. (1998). Cell, 92, 351–366. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Hartl, F. U. (1996). Nature (London), 381, 571–579. [DOI] [PubMed]
- Hollien, J. & Weissman, J. S. (2006). Science, 313, 104–107. [DOI] [PubMed]
- Huizen, R. van, Martindale, J. L., Gorospe, M. & Holbrook, N. J. (2003). J. Biol. Chem.278, 15558–15564. [DOI] [PubMed]
- Kang, S. W., Rane, N. S., Kim, S. J., Garrison, J. L., Taunton, J. & Hegde, R. S. (2006). Cell, 127, 999–1013. [DOI] [PMC free article] [PubMed]
- Ladiges, W. C., Knoblaugh, S. E., Morton, J. F., Korth, M. J., Sopher, B. L., Baskin, C. R., MacAuley, A., Goodman, A. G., LeBoeuf, R. C. & Katze, M. G. (2005). Diabetes, 54, 1074–1081. [DOI] [PubMed]
- Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., Shokat, K. M., Lavail, M. M. & Walter, P. (2007). Science, 318, 944–949. [DOI] [PMC free article] [PubMed]
- Malhotra, J. D. & Kaufman, R. J. (2007). Semin. Cell Dev. Biol.18, 716–731. [DOI] [PMC free article] [PubMed]
- Marciniak, S. J., Garcia-Bonilla, L., Hu, J., Harding, H. P. & Ron, D. (2006). J. Cell Biol.172, 201–209. [DOI] [PMC free article] [PubMed]
- Marciniak, S. J. & Ron, D. (2006). Physiol. Rev.86, 1133–1149. [DOI] [PubMed]
- Melville, M. W., Tan, S. L., Wambach, M., Song, J., Morimoto, R. I. & Katze, M. G. (1999). J. Biol. Chem.274, 3797–3803. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Oyadomari, S., Yun, C., Fisher, E. A., Kreglinger, N., Kreibich, G., Oyadomari, M., Harding, H. P., Goodman, A. G., Harant, H., Garrison, J. L., Taunton, J., Katze, M. G. & Ron, D. (2006). Cell, 126, 727–739. [DOI] [PubMed]
- Ron, D. & Harding, H. P. (2007). Translational Control in Biology and Medicine, edited by M. B. Matthews, N. Sonenberg & J. W. B. Hershey, pp. 345–368. New York: Cold Spring Harbor Laboratory Press.
- Ron, D. & Walter, P. (2007). Nature Rev. Mol. Cell Biol.8, 519–529. [DOI] [PubMed]
- Rutkowski, D. T., Kang, S. W., Goodman, A. G., Garrison, J. L., Taunton, J., Katze, M. G., Kaufman, R. J. & Hegde, R. S. (2007). Mol. Biol. Cell, 18, 3681–3691. [DOI] [PMC free article] [PubMed]
- Rutkowski, D. T. & Kaufman, R. J. (2007). Trends Biochem. Sci.32, 469–476. [DOI] [PubMed]
- Scheufler, C., Brinker, A., Bourenkov, G., Pegoraro, S., Moroder, L., Bartunik, H., Hartl, F. U. & Moarefi, I. (2000). Cell, 101, 199–210. [DOI] [PubMed]
- Schroder, M. & Kaufman, R. J. (2005). Annu. Rev. Biochem.74, 739–789. [DOI] [PubMed]
- Wu, J., Rutkowski, D. T., Dubois, M., Swathirajan, J., Saunders, T., Wang, J., Song, B., Yau, G. D. & Kaufman, R. J. (2007). Dev. Cell, 13, 351–364. [DOI] [PubMed]
- Yan, W., Frank, C. L., Korth, M. J., Sopher, B. L., Novoa, I., Ron, D. & Katze, M. G. (2002). Proc. Natl Acad. Sci. USA, 99, 15920–15925. [DOI] [PMC free article] [PubMed]
- Young, J. C., Hoogenraad, N. J. & Hartl, F. U. (2003). Cell, 112, 41–50. [DOI] [PubMed]