

Abstract
DF1 is a small, idealized model for carboxylate-bridged diiron proteins. This protein was designed to form a dimeric four-helix bundle with a dimetal ion-binding site near the center of the structure, and its crystal structure has confirmed that it adopts the intended conformation. However, the protein showed limited solubility in aqueous buffer, and access to its active site was blocked by two hydrophobic side chains. The sequence of DF1 has now been modified to provide a very soluble protein (DF2) that binds metal ions in a rapid and reversible manner. Furthermore, the DF2 protein shows significant ferroxidase activity, suggesting that its dimetal center is accessible to oxygen. The affinity of DF2 for various first-row divalent cations deviates from the Irving–Willliams series, suggesting that its structure imparts significant geometric preferences on the metal ion-binding site. Furthermore, in the absence of metal ions, the protein folds into a dimer with concomitant binding of two protons. The uptake of two protons is expected if the structure of the apo-protein is similar to that of the crystal structure of dizinc DF1. Thus, this result suggests that the active site of DF2 is retained in the absence of metal ions.
Keywords: De novo protein design, four-helix bundle, diiron, metalloprotein design
Proteins use a limited set of metal ions to mediate a variety of processes. Often the same metal center can serve a number of roles; for example, binuclear iron centers engage in reversible binding of oxygen, electron transfer, and the catalysis of hydroxylation reactions (Feig and Lippard 1994; Andersson and Gräslund 1995; Nordlund and Eklund 1995; Waller and Lipscomb 1996; Lange and Que 1998). How a protein is able to fine-tune the properties of a metal ion to effect such different functions is an important question that may now be addressed by de novo protein design (Regan 1995; Hellinga 1998). Metal-binding sites in proteins have classically been assigned as serving either structural or functional roles (da Silva and Williams 1993). Structural sites generally stabilize or direct the folding of the protein and exhibit common, coordinately saturated geometries that are well precedented in simple small-molecule/metal-ion complexes. By contrast, functional metal-binding sites often show fine-tuned ligation geometries, which allow rapid electron transfer and/or binding of substrates.
Initial attempts at metalloprotein design focused on engineering structural zinc-binding sites (Handel and DeGrado 1990; Regan and Clarke 1990; Handel et al. 1993; Pessi et al. 1993). These exercises met with considerable success, leading to attempts to design functional metal-binding sites. For example, models for rubredoxin, ferredoxin, and heme proteins have been reported (Choma et al. 1994; Robertson et al. 1994; Gibney et al. 1996; Rabanal et al. 1996; Benson et al. 1998). In addition, a mononuclear iron site has been introduced into thioredoxin, resulting in a protein with superoxide dismutase activity (Pinto et al. 1997). Recently we described the design and structure determination of Due Ferro 1 (DF1), a carboxylate-bridged diiron protein that mimics the structures of the active sites of methane monooxygenase, the R2 subunit of ribonucleotide reductase, stearoyl-ACP Δ9 desaturase, and the ferritins (Lombardi et al. 2000).
DF1 is a homodimeric protein whose structure idealizes the quasi-twofold symmetry observed in natural diiron proteins. The dimer consists of two identical helix–loop–helix motifs that assemble about a twofold axis directed through the center of the metal-binding site. The crystal structure (Fig. 1 ▶) contains two zinc ions in the active site, the structure of which is very close to that found in the diferrous and dimanganous forms of naturally occurring proteins (Frolow et al. 1994; Lindqvist et al. 1996; Andersson et al. 1999). Each metal ion is 5-coordinate, with two Glu carboxylates, each interacting with both zinc ions in a 1,3 syn–syn bidentate bridging interaction, and two Glu side chains, each binding individual ions in a chelating manner. Two His residues complete the ligation environment about the dimetal site, by Nδ atom coordination to individual ions. The Zn2+–Zn2+ distance is 3.9 Å, close to the distance observed between metal ions in the dimanganous and diferrous forms of natural diiron proteins. The pentacoordinate ligand arrangement with two 1,3 bridging carboxylates and two chelating carboxylates is also nearly identical to that observed in diferrous Δ9-ACP desaturase (Lindqvist et al. 1996) and di-Mn2+ bacterioferritin (Frolow et al. 1994). The ligands surround the metal ions except for a vacant pair of sites along one face of the dimetal center. The vacant sites are oriented trans to the His ligands, and are well oriented for interaction with bridging ligands. If the diiron site of DF1 adopts a geometry similar to that of the di-Zn2+ structure, it should be well suited for reaction with O2.
Fig. 1.

The crystal structure of DF1. Perpendicular views of the crystal structure are shown. The protein is a homodimer; the monomers are shown in different colors. The two spheres are zinc ions. The zinc ligands are shown, and consist of one terminally chelating Glu residue from the first helix of each monomer, and one bridging Glu and one His from the Glu–X–X–His motif in the second helix of each monomer. In the right-hand picture, the helices are clipped for an unobstructed view of the metal-binding site. The two Leu 13 residues (position 14 in DF2) block access to the dimetal site, and have been replaced in DF2 with Ala.
An important question for the design of metalloproteins is the extent to which a protein structure influences the ligand geometry, metal-binding specificity, and reactivity of a metal-binding site. To address this question, we used a recombinant version of DF1 that additionally has several changes in the sequence improving its water solubility and the accessibility of oxygen to its dimetal site. We measured the affinity of this new protein, DF2, for the series of first-row divalent transition metal ions from Mn2+ to Zn2+. Furthermore, to determine the extent to which the metal ions influence the stability of the structure, we examined the folding of the protein in the absence of metal ions. In the apo state the buried Glu ligands would be expected to destabilize the folded dimer electrostatically, possibly resulting in protonation upon folding. If this were the case, one would expect the free energy of dimerization to become increasingly less favorable as the pH is increased over the pKa of the Glu side chains in the unfolded monomers. Therefore, we examined the pH dependence of the dimerization of this peptide. Finally, we examined the reactivity of the ferrous form of the protein toward O2.
Results
Design of DF2
Although the initial design of DF1 was successful in that it adopted the intended structure and bound two metal ions, certain limitations hindered future progress toward the design of catalytically active variants of DF1. One limitation was that the water solubility of DF1 was poor, limiting its concentration in aqueous solution (in the absence of organic solvent) to approximately 10 μM. Upon reexamination of the DF1 model, we decided that several hydrophobic residues at interfacial, partially solvent-exposed positions would be amenable to mutation to more hydrophilic residues in order to increase its water solubility. Most changes to the DF1 sequence were introduced for this purpose (Fig. 2 ▶), and, indeed, the new peptide, DF2, is more water-soluble. Leu residues at positions 7, 11, 13, 21, 26, and 47 were changed to Tyr, Gln, Ala, Ser, Asp, and Asn, respectively. These side chains were chosen to increase both the hydrophilicity and also the sequence diversity for future NMR studies. Lys 25 in the turn was changed to Gly to stabilize an αL conformation, which was necessary for a Schellman motif (Schellman 1980). The Q17L mutation arose from an error in a PCR primer, but was retained when it was found to stabilize the structure. Depending on the pH used (DF2 solubility increases as pH is increased over the range of 4.0–7.0), DF2 is soluble up to at least 0.5 mM.
Fig. 2.

Sequence changes in DF2 relative to DF1. Sequence changes are in boldface. Several changes were made to increase solubility. A Met is the first residue in DF2 in order to express DF2 in Escherichia coli. Leu 13 in DF1 was replaced with Ala 14 in DF2 to increase accessibility to the dimetal site. The Gln 16 to Leu 17 change was necessary for efficient protein expression. The turn residues were replaced with –GlyAsp– in DF2 because these were found to be the most common amino acids at their respective positions in a two-residue turn between two helices, as determined by a database search using the program WHATIF (Vriend 1990).
An additional problem that has been addressed in DF2 is that the metal-binding site of DF1 was relatively inaccessible. Metal ions bound extremely slowly unless the protein was first denatured and then refolded in the presence of metal. Also, DF1 complexed with iron did not readily form a diferric oxo-bridged species, contrary to results presented here for DF2, suggesting that either oxygen could not access the metal site or that the more hydrophobic site in DF1 disfavors oxidation states higher than Fe2+. This is prohibitive toward the goal of developing catalytically active versions of DF1. The crystal structure of DF1 clearly showed that Leu 13 (position 14 in DF2) blocked access of the metal center to oxygen or other substrates (Fig. 1 ▶). Therefore, this residue has been replaced with Ala, creating a cavity adjacent to the dimetal site in the dimer. Finally, whereas DF1 had been chemically synthesized, DF2 has been expressed in Escherichia coli (see Materials and Methods) to facilitate the economic preparation of mutants as well as biosynthetic labeling for NMR studies.
Oligomeric state of DF2
The primary concern arising from the many changes in DF2 relative to DF1 was whether the new molecule possessed sufficient hydrophobic stabilization to maintain the designed dimeric structure. Therefore, analytical ultracentrifugation was used to verify whether DF2 forms a stable dimer, both in the absence and presence of metal ions. The oligomeric state of apo-DF2 is pH-dependent (Fig. 3 ▶; Table 1). The data at pH 4.0, pH 6.0 (Fig. 3a ▶), and pH 6.5 (Fig. 3b ▶) were analyzed using a model with a single molecular species. This model fit the pH 4.0 data well, returning a molecular weight of 11,610, consistent with fully dimeric DF2 (theoretical weight 11,790). However, the data at both pH 6.0 and pH 6.5 yielded a molecular weight between that expected for a dimer and a trimer. This finding suggested that at near neutral pH in the absence of metal ions, DF2 formed higher-order aggregates. Therefore, curves expressing a cooperative monomer–trimer equilibrium and a monomer–dimer–trimer equilibrium were fitted to the data. The data were well described by a noncooperative monomer–dimer–trimer equilibrium shown in Figure 3 ▶, a (pH 6.0) and b (pH 6.5). As expected, the formation of the higher-order aggregate is favored at higher peptide concentrations and at higher pH. For example, at a relatively high concentration of 100 μM, DF2 is essentially fully dimeric at pH 4.0. However, at this protein concentration, as the pH is increased the population of oligomers increases until they predominate at pH 6.5. Thus as the pH increases, the stability of the dimer decreases until it is comparable to the alternately folded oligomers.
Fig. 3.



Equilibrium analytical ultracentrifugation of DF2 at different pH values and in the presence and absence of metal ions. Solutions of 100 μM DF2 in the appropriate buffer for each pH were centrifuged at 40,000 rpm. Absorption measurements were made after sedimentation equilibrium was reached. Each panel shows data fitted to a monomer–dimer–trimer equilibrium scheme. In each panel, residuals are shown on top and the species plot on the bottom. (a) DF2 at pH 6.0. (b) DF2 at pH 6.5. (c) DF2 at pH 6.0 with 100 μM Zn.
Table 1.
Fractions of DF2 dimer and trimer DF2 from analytical ultracentrifugation
| Experiment | Fraction dimer | Fraction trimer |
| pH 4.0 | 0.81–0.96 | — |
| pH 6.0 | 0.32–0.71 | 0.22–0.68 |
| pH 6.5 | 0.07–0.22 | 0.68–0.92 |
| pH 6.0 Zn | 0.97–0.99 | <0.003 |
Ultracentrifugation was also performed on DF2 in the presence of metal ions (Fig. 3c ▶) to determine whether they would specifically stabilize the dimeric state. Importantly, at pH 6.0, 100 μM apo-DF2 in the presence of 100 μM Zn2+ ions is entirely dimeric. Furthermore, DF2 (100 μM) was reconstituted at pH 6.5 with 2 equivalents Fe2+/dimer under aerobic conditions (see below), and subjected to analytical ultracentrifugation. The data were well described by a unimolecular species with an apparent molecular mass (10,540 D) in good agreement with that expected for the reconstituted dimer (11,902 D). Thus, the binding of metal ions specifically stabilizes the desired dimer relative to alternately folded states.
DF2 stability is pH-dependent; two protons are added to DF2 upon folding
The thermodynamic stability of DF2 in the absence of metal is expected to be pH-dependent, as a number of the glutamic acid metal ligands may become protonated upon folding. Denaturation curves of DF2 measured at different pHs illustrate that its stability is, indeed, pH-dependent (Fig. 4a ▶). Through the pH range measured (pH 4–7), DF2 is least stable at pH 7 and its stability increases with decreasing pH. The denaturation curves also depend on the concentration of DF2, which indicates an equilibrium between unfolded monomers and structured dimers (or higher-order oligomers at high pH). The number of protons taken up by DF2 upon folding are determined from these data using equation 1:
Fig. 4.


pH-dependent denaturation of DF2. (a) Stability of DF2 as a function of pH was determined by guanidine denaturation curves measured by CD. Open symbols designate a DF2 concentration of 10.7 μM, and filled symbols are for a DF2 concentration of 33.3 μM. (open diamond) pH 7.0, (closed circle) pH 6.5, (open triangle, point down) pH 6.0, (open square) pH 5.5, (open and closed triangles, point up) pH 5.0, (open circle) pH 4.0. Curves were determined using pH 4.0, pH 5.0, and pH 5.5 data assuming two-state denaturation from dimer to monomer, because trimer concentration at these pH values is small. Dashed line curves are theoretical curves for pH 6.0, pH 6.5, and pH 7.0 predicted from the lower pH data. The calculated curve for pH 6.0 deviates slightly from the experimental data, and the deviation between the curves and the data becomes progressively larger as the pH is raised. (b) DF2 takes up two protons upon folding. The negative of the slope of the plot (Eq. 2 in Results) is the proton uptake, equal to 1.9 ± 0.2. Observed equilibrium constants were determined from curves in a. Higher pH data were not used because of the significant trimer population, and pH 4.0 data were not used because Glu residues would be expected to be protonated in the unfolded state.
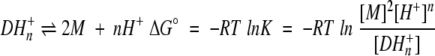 |
1 |
in which M is monomeric (unfolded) DF2, D is dimeric (folded) DF2, and K is the dissociation constant including the uptake of protons. At a given pH, an observed equilibrium constant, Kobs, could be expressed in the form:
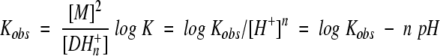 |
2 |
allowing the proton uptake n to be determined from a plot of log Kobs versus pH (Fig. 4b ▶). Only data from pH 5.0–6.0 were used for this analysis to avoid potential complications associated with the formation of trimers and higher-order oligomers at higher pH. The resulting slope of −1.9 ± 0.2 indicates that two protons are taken up upon forming the folded dimer.
The observation that two protons are required for DF2 to fold, together with the crystallographic structure of DF1, suggests a mechanism for oligomerization. In the structure of dimeric DF1, four Glu and two His side chains are fully or partially buried. This cluster may be expected to be present in an ionization state that is overall neutral, as has been observed in apo-bacterioferritin (Le Brun et al. 1996; Keech et al. 1997) and inferred from the crystal structure of the iron-free R2 subunit of ribonucleotide reductase, in which short distances were observed between the buried carboxyl groups (Aberg et al. 1993). Electrical neutrality is satisfied when all four Glu residues are protonated and the two His residues are deprotonated (or in other forms with equivalent numbers of protons; e.g., with two Glu residues in a neutral protonated form, two Glu residues negatively charged, and two His residues in the positively charged, protonated forms). These protonation states differ by approximately two protons relative to the predominant protonation state of two DF2 monomers between pH 4.5 and 6, in which the Glu side chains would be largely deprotonated and the His residues would be protonated, explaining the net uptake of two protons. Thus, the data were analyzed using scheme 1 ▶, which describes the protonation of the monomer (in which MH3+, MH2, MH−, and M2− are the triply protonated through the unprotonated forms of the liganding amino acids of the monomer, respectively), under the assumption that only the neutral form, MH2, is able to dimerize. Furthermore, because the oligomers are formed only at elevated pH, this scheme assigns trimer formation to the negatively charged species, MH−.
Scheme 1.
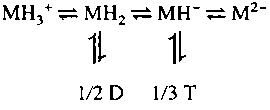
We wished to determine the pH range over which the presence of oligomer is sufficiently small that the association may be approximated as a simplified form of scheme 1 ▶, that would not allow the formation of the trimeric species, T, or other higher-order aggregates. Therefore, curves representing a monomer–dimer equilibrium, with the proton uptake fixed to exactly two protons (Fig. 4a ▶), were fitted only to data between pH 4.0 and pH 5.5, where the concentration of trimers would be negligible as assessed by sedimentation equilibrium. The curves fit extremely well to the data in this range of pH, providing strong support for the scheme, as well as for the assumption that two protons are bound upon dimerization. The parameters were then used to create theoretical curves for the data at pH 6.0, pH 6.5, and pH 7.0 (the dashed lines in Fig. 4a ▶). Therefore, deviations of these lines from the data may be associated with the presence of oligomers. The data at pH 6.0 are well described by this scheme, and the deviation is close to that expected from experimental error. However, the fit of the curves to the data becomes increasingly poor as the pH is increased. We therefore chose pH 6.0 as a convenient condition to measure binding of metal ions. At this pH the solubility of divalent transition metals is good, and the protein is predominantly dimeric at relatively low peptide concentration.
Two metal ions bind to and stabilize DF2
Although DF2 is marginally stable at pH 6.0 in the apo state, it should be stabilized upon specifically binding divalent cations. We measured guanidine denaturation curves for 2–3 μM DF2 at pH 6.0 with varying metal ion concentrations (in excess of protein) to determine the stoichiometry and overall binding affinity. Because the denaturation curve of 11 μM DF2 at pH 6.0 is reasonably well fit by a monomer–dimer equilibrium (Fig. 4a ▶), the lower DF2 concentration in the metal binding experiments indicates that any residual contribution of oligomers would be further minimized. Addition of Zn2+ ions to DF2 at pH 6.0 is greatly stabilizing (Fig. 5 ▶). Analogous to the determination of number of protons bound, the number of metal ions bound was determined from a plot of the logarithm of the dimerization constant versus the logarithm of the metal ion concentration (data not shown). The slope of −1.8 indicates that DF2 binds two zinc ions per dimer.
Fig. 5.


The effect of M2+ ions on DF2 stability. (a) Solutions of 2.8 μM DF2 at pH 6.0 in varying amounts of GdnHCl were incubated overnight with different amounts of ZnCl2. (closed circle) 20 μM, (closed square) 50 μM, (closed triangle, point up) 100 μM, (closed triangle, point down) 200 μM. Dashed line fit is for DF2 with no metal. (b) Solutions of 2.5 μM DF2 at pH 6.0 in varying amounts of GdnHCl were incubated overnight with different amounts of MnCl2. (closed triangle, point up) 100 μM, (closed triangle, point down) 200 μM, (closed circle) 400 μM, (closed square) 1000 μM. Dashed line fit is for DF2 with no metal.
The binding stoichiometry was confirmed by a direct titration experiment with Co2+. In aqueous solution, Co2+ is octahedrally coordinated, and hence shows very weak d–d electronic transitions. However, upon binding in a pentacoordinate geometry to DF2, one would expect a large increase in its extinction coefficient (Bertini and Luchinat 1984). Indeed, the spectrum of Co2+ in the presence of stoichiometric amounts of DF2 is virtually indistinguishable from Co2+-reconstituted bacterioferritin (Fig. 6a ▶; Keech et al. 1997). The coordination geometry in the di-Mn2+-substituted form of bacterioferritin is pentacoordinate and virtually identical to that of di-Zn2+-substituted DF1 (Frolow et al. 1994). The large increase in extinction coefficient can be used to quantify the extent of binding in titration experiments. The titration of 70 μM DF2 with Co2+ shows that the curve abruptly levels off at a stoichiometry of two Co2+ per dimer (Fig. 6b ▶). Therefore, two Co2+ ions are bound to DF2, in agreement with the Zn2+-binding experiment as well as the crystal structure of DF1. The sharpness of the transition indicates that the average dissociation constant is more favorable than ∼20 μΜ.
Fig. 6.


(a) The visible absorption spectrum of Co2+ when bound to DF2. Peaks occur at 520, 550, and 600 nm. Both DF2 and Co2+ concentrations are 70 μM. The extinction coefficient (Y axis) is per Co2+ ion. (b) Titration of DF2 with Co2+ monitored by visible absorption at 550 nm. DF2 concentration was 70 μM. No further increase in absorption is observed after two equivalents of Co2+ per DF2 are added, indicating that two Co2+ ions bind per DF2 dimer. The line is a curve of best fit generated with a dissociation constant of 0.6 μM.
Transition metal binding to DF2 shows deviations from the Irving–Williams series
Binding constants for DF2 with Mn2+, Co2+, Ni2+, and Cu2+ were determined similarly to that for Zn2+ (Table 2). Similar relative affinities of bacterioferritins for Zn2+, Co2+, and Fe2+ have been reported (Le Brun et al. 1996; Yang et al. 2000). Data for Mn2+, the most weakly binding ion, are also shown in Figure 5b ▶. Denaturation of DF2 in the presence of Cu2+ shows that DF2 binds to and is stabilized by Cu2+ at very low Cu2+ concentrations (Fig. 7 ▶). However, further increases in the Cu2+ concentration have a destabilizing effect, suggesting that there are a number of lower-affinity Cu2+ binding sites in the unfolded protein. The coordination in the unfolded state may involve backbone amide groups, which are known to coordinate Cu2+ (Hay et al. 1993; Brittain et al. 1998; Torrado et al. 1998; Stemmler and Burrows 1999). Of the first-row transition metal ions tested, Ni2+ was the only one that did not stabilize DF2 at any concentration ranging from 20 to 2000 μM (not shown).
Table 2.
Metal ion association constants
| Metal ion | pKmet |
| Zn | 6.73 |
| Co | 4.81 |
| Mn | 4.08 |
| Cu | 5.79 |
| Fe | 4.75 |
Fig. 7.

The effect of Cu2+ ions on DF2 stability. Data are shown for (closed square) 20 μM Cu2+ and (closed triangle) 40 μM Cu2+, along with their fit to the model discussed in Materials and Methods. Data are also shown for 200 μM Cu2+ (open triangles) and 400 μM Cu2+ (open circles). These data show that Cu2+, although stabilizing at low concentrations, becomes destabilizing at the higher concentrations. The leftmost curve is the fit for no metal.
Formation constants for first-row transition metal complexes with small ligands generally follow the Irving–Williams series (da Silva and Williams 1993): Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+. Thus, Cu2+ complexes are generally the most stable, and Ni2+ complexes are also relatively tight. This order is contrary to what we observe in DF2 (Table 2), where formation constants for transition metal binding to DF2 do not follow the Irving–Williams series, as Cu2+ binds less tightly than Zn2+, and Ni2+ binds the weakest of all the transition metals measured, orders of magnitude weaker than expected. Such deviations from the Irving–Williams series indicate that in its folded state, the metal-binding site of DF2 has limited flexibility, and imparts geometric specificity to the arrangement of the ligands. Nickel and copper often form complexes of square planar geometry (Huheey et al. 1993). The observation that these metals bind to DF2 more weakly than expected from the Irving–Williams series implies that the active site of DF2 is not sufficiently flexible to accommodate the preferred square planar geometry.
DF2-catalyzed oxidation of Fe2+
Many diiron proteins catalyze the reaction of O2 with ferrous ions to provide oxo-bridged diferric protein. To determine the reactivity of the diferrous center in DF2, we used assay conditions that have been extensively employed to measure the reactivity of the ferritins and bacterioferritins (Yang et al. 1998, 2000). Fe2+ was added to protein under aerobic conditions, and the extent of oxidation was measured by monitoring the appearance of a charge transfer band associated with the oxo-bridged diferric species. Similarly, the addition of Fe2+ to DF2 under aerobic conditions results in a time-dependent formation of a UV-visible spectrum that is identical to the spectrum previously reported for the oxo-bridged diferric form of bacterioferritin (Fig. 8a ▶).
Fig. 8.



The time dependence of the increase in absorbance following incubation of Fe2+ with DF2 (83 μM protein concentration; 015 M NaCl, 0.15 M MES at pH 6.5, 25°C). Panel a illustrates stacked spectra taken at various times (6 min, 11 min, 16 min, 21 min, 26 min, and 31 min). Panel b illustrates the variation in rate with respect to protein concentration (constant Fe2+ = 83 μM). Panel c illustrates the variation in rate with respect to Fe2+ concentration (constant protein concentration = 83 μM).
The addition of DF2 results in a substantial increase in the rate of iron oxidation, relative to the background reaction. Figure 8, b and c ▶, illustrates the effect of varying the concentration of protein and Fe2+ on the initial rate of iron oxidation. The reaction is first order in both iron and protein. Furthermore, experiments in which protein was incubated for long times with Fe2+ indicate that the resulting Fe(III) species is stable and remains associated with DF2 for several hours. To determine the extinction coefficient for the oxidized iron species, the protein was allowed to react with 3.0 equivalents of Fe2+ per dimer for 1 h, and then chromatographed through a size exclusion column. A major peak was observed at the position expected for the dimer, and quantification of the iron and protein content indicated 2.0 equivalents metal ion per dimer. The extinction coefficient for the diiron center at 300 nm, 3240 M−1 cm−1 (on a per iron basis) was in excellent agreement with the value of 3380 M−1 cm−1 previously reported for the ferroxidase site of bacterioferritin (Yang et al. 2000), and comparable to the extinction coefficients for human H-chain ferritin, 2990 M−1 cm−1, and horse spleen ferritin, 3540 M−1 cm−1 (Yang et al. 1998). The extinction coefficient for the DF2 diiron center at 325 nm, 2400 M−1 cm−1, compares with that of other diiron proteins: ribonucleotide reductase, 4700 M−1 cm−1 (Bollinger et al. 1991) and stearoyl-ACP Δ9d-desaturase, 2080 M−1 cm−1 (Fox et al. 1994); as well as with a carboxylate/oxo-bridged model complex, 3400 M−1 cm−1 (Tolman et al. 1991).
The rate of oxidation of Fe2+ in the presence of DF2 is approximately three orders of magnitude slower than that for bacterioferritin. To determine whether the chemical step is rate-determining, we preincubated DF2 with a stoichiometric amount of Fe2+ under anaerobic conditions for 45 min. The resulting complex was then diluted twofold with aerated buffer, which resulted in the quantitative oxidation of the Fe2+ center within 30 sec as assessed by a rapid increase in absorbance of the ligand-to-metal charge transfer band. Therefore, the oxidation step is not rate-determining; rather, the initial binding of Fe2+ or a subsequent conformational change appears to limit the rate of reaction with O2.
Discussion
In this paper we describe an important step in the de novo design of metalloproteins. The design of proteins with precisely predetermined substrate-binding pockets, capable of binding substrates proximal to a redox-active cofactor, has been a difficult undertaking. By modifying the sequence of DF1, we have now shown that it is possible to accomplish this objective. DF2 binds metal ions rapidly, and also displays ferroxidase activity, strongly suggesting that the active site is accessible to oxygen. The rate of oxidation of iron by DF2 is within two to three orders of that observed in ferritins and bacterioferritin (Yang et al. 2000), and it will be of considerable interest to determine how additional modifications to the sequence modulate the rate.
Another major issue explored in this paper is the interplay between protein folding and metal–ligand interactions in defining the detailed geometry of the active site of DF2. The affinities of DF2 for various first-row divalent cations deviates from the Irving–Williams series, indicating that its structure imparts significant geometric preferences on the metal ion-binding site. Similar results have been described for three single-iron-binding sites designed on a thioredoxin scaffold, in which no nickel binding was observed (Benson et al. 2000). Furthermore, in the absence of metal ions, the protein folds into a dimer with concomitant binding of two protons. The uptake of two protons is to be expected if the structure of the apo-protein is similar to that of the crystal structure of di-zinc DF1. This result therefore suggests that the active site of DF2 is retained in the absence of metal ions. Interestingly, as the pH is increased the energetic cost of protonating the Glu side chains in the active site becomes increasingly unfavorable, until the protein aggregates near neutral pH. In this respect, DF2 is not as highly evolved as naturally occurring diiron proteins, whose apo forms are generally stable at neutral pH. However, for many applications this should not be a problem, because DF2 forms the desired dimer when it binds its dimetal ion cofactor. Thus, DF2 provides an attractive platform for examining the relationship between amino acid sequence and cofactor reactivity in diiron proteins.
Materials and methods
DF2 gene construction and DF2 expression and purification
A synthetic gene for DF2 was constructed using PCR from four oligonucleotides as described by Dillon and Rosen (1993). The sequence of the full gene is: atggactacctgcgtgaactgtacaaac tggaacagcaggcgatgaaactgtaccgtgaagcttctgaacgtgttggtgacccggttctgg ctaaaatcctggaagacgaagaaaaacacatcgaatggctggaaactatcaacggt. The DNA sequence design included a few more bases on either side of the gene containing restriction sites for enzymes NcoI and BamHI. Following digestion with these enzymes, the gene and the pET-15b (Novagen) plasmid were ligated together. The pET-15b vector containing the DF2 gene was then transformed into BL21-Dɛ3 cells for expression. Protein expression was induced with 1 mM IPTG when cell growth in LB media reached OD600 = 0.6. Maximum expression occurred within 1 h following induction. Cells were pelleted and sonicated in cold 10 mM Tris at pH 7.5. After centrifugation, the crude cell lysate was heated to 80°C for 10 min, and denatured proteins were pelleted. Most of the DF2 remained soluble, and thus the heating step served as the initial purification. DF2 was further purified by reverse-phase HPLC on a C4 column. The protein molecular weight was determined with MALDI mass spectroscopy, which indicated that the Met N terminus was intact.
Analytical ultracentrifugation
Sedimentation equilibrium experiments were performed at 25°C using a Beckman XL-I analytical ultracentrifuge (Furst 1997). The peptide monomer molecular mass, partial specific volume, and molar extinction coefficient were calculated using the program SEDINTERP (Laue et al. 1992), modified to include the amino acid residue partial specific volumes reported by Kharazov (Kharakoz 1997). These values were fixed to be constants in fitting the radial concentration profiles to different equilibrium association models. The samples (100 μM DF2 monomer in 50 mM NaAc/100 mM NaCl at pH 4.0, and 50 mM MES/100 mM NaCl at pH 6.0 and pH 6.5) were centrifuged to equilibrium at 45,000 rpm in an An50Ti rotor employing three-compartment carbon-epoxy centerpieces with quartz windows. Data obtained by UV absorption at 290 nm were analyzed by nonlinear least squares curve fitting of radial concentration profiles using the Marquardt–Levenberg algorithm implemented in Igor Pro (Wavemetrics). Each data set was fitted to a monomer–dimer–trimer reversible equilibrium model with logarithmic dissociation constants as fitting parameters. Baselines and signal values at the meniscus were allowed to vary in fitting. After fitting, the contributions of each species to the signal were calculated as a function of the concentration as measured from the baseline-corrected UV absorbance signal. The upper and lower limits of the peptide fraction of each species were calculated and tabulated along with the fitted values of the negative logarithm (pK) of the dissociation constants for each experiment (Table 1).
Guanidine denaturation curves
The stability of DF2 was evaluated as a function of pH, peptide concentration, and various metal ion concentrations, by monitoring the CD signal at 222 nm versus the concentration of guanidine. Solutions of 2–33 μM DF2 in 50 mM NaAc (for pH 4.0, pH 5.5), MES (for pH 6.0, pH 6.5), or MOPS (for pH 7.0) with 100 mM NaCl and GdnHCl varying from 0 M to 5 M, were prepared and allowed to equilibrate overnight because the folding/unfolding was observed to be slow, before measuring the CD signal at 222 nm.
Binding constants of divalent transition metal ions to DF2 were measurable by guanidine denaturation, because metals stabilize DF2 in a concentration-dependent manner. Solutions were left to equilibrate overnight. As complexes of Ni2+ are often slow to exchange, the Ni2+ solutions were measured a second time after five days. With all metals, the DF2 concentration was 2–3 μM. For each metal except iron, curves of typically eight guanidine concentrations were measured at four different metal concentrations ranging from 20 μM to 2000 μM, depending on the metal. In the iron experiment, five data points at only one iron concentration, 200 μM, were measured. Solutions were deoxygenated with argon and left overnight in a glovebox before mixing protein/guanidine and iron solutions, after which they were left again overnight, along with the cuvets, to equilibrate. The following morning the solutions (in sealed cuvets) were removed from the glovebox, and their CD signals were promptly measured.
The data were analyzed assuming that the measured signal of each solution was the sum of contributions from the unfolded monomer, [θ]u, and from the folded, [θ]f, helical dimers, trimers, and metal-bound dimers, with no distinction made among the folded dimeric and trimeric species. Baselines representing signals from pure unfolded and folded species were assumed to be linear functions of guanidine concentration and were included as fitting parameters. The function used to fit the data was:
 |
3 |
 |
4 |
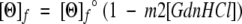 |
5 |
with fm, the fraction of monomer, computed from the equilibrium relationships discussed below.
The presence of glutamic acid and histidine residues in the peptide, together with the pH dependence of the ultracentrifuge data, suggested the following equilibrium scheme:
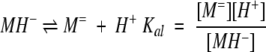 |
 |
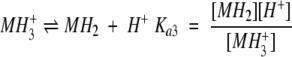 |
 |
 |
 |
where M= is the monomer with two deprotonated Glu and one deprotonated His residue; MH− is the monomer with two deprotonated Glu and one protonated His residue; MH2 is the monomer with one deprotonated Glu, one protonated Glu, and one protonated His residue; MH3+ is the fully protonated monomer; D is the dimer formed from two MH2 monomers; T is the trimer formed from three MH− monomers; X is a metal ion; C is the complex formed by nmet metal ions and the dimer; and the K's are defined equilibrium constants.
The choice of the MH− species as the trimerization-competent monomer is suggested by the analytical ultracentrifugation data, which showed trimerization to be more extensive at pH 6.5 than at pH 6.0, indicating that the trimer is formed from a more negatively charged species.
The equilibrium equations were solved for the fraction of total peptide that is present at equilibrium as a monomer (fm) by using the material balance condition to solve for one of the species (here, MH2):
 |
Dividing through by Pt and collecting terms gives:
 |
This equation was solved for z, the fraction of total peptide present as MH2 monomer, using an iterative root-finding algorithm in IgorPro (Wavemetrics). Finally, fm, the total monomer fraction, was calculated by adding the fractions for M=, MH−, and MH2+.
Kd and Kt were assumed to vary linearly with guanidine hydrochloride (GdnHCl) concentration according to the following expressions:
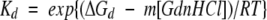 |
 |
In these equations it is assumed that m is proportional to the solvent-accessible surface area and therefore that the trimer has 1.5 times the value of the monomer.
The parameters describing the dependence of the monomer and dimer ellipticities and the dimer thermodynamic stability on the GdnHCl concentration were determined by curve fitting. These parameters were assumed to be independent of pH and of metal ion concentration (were a global fitting parameter for datasets of different pH values and for datasets of different metal ion concentrations), but were allowed to differ for each type of metal ion. The use of the equations above resulted in a reasonable fit to the data up to pH 6.0, and produced a value for the dimerization constant Kd of 2.4 × 10−11 M−1. In subsequent experiments, the contribution from the trimeric species was eliminated by fixing Kt to a very high value, to determine the pH range over which more a simple, pH-dependent monomer–dimer equilibrium adequately described the data. In fitting curves to the metal-binding data, the dimerization constant was fixed at 2.4 × 10−11 M−1. After determining the number of metal ions bound to be 2, this parameter was fixed for the determination of the metal-binding constants.
Oxidation of Fe2+-reconstituted DF2
Kinetic experiments were performed under aerobic conditions at pH 6.5 (0.15 M MES/0.15 M NaCl). To each DF2 solution (70 μM) was added varying amounts of a solution of 1 mM ferrous ammonium sulfate/0.01% sulfuric acid. For samples with varying protein concentration, the ferrous ion concentration was held at 70 μM. The sample was mixed for 30 sec, and spectra were measured on a Hewlett Packard diode array spectrophotometer. Spectra were collected each in 0.5–1.0 min increments for 15 min. The initial rate was measured at 320 nm from points at which less than 10% of the Fe2+ had been consumed (generally less than 4 min). At longer times, small amounts of light-scattering material began to form and precluded accurate kinetic measurements.
To determine the stoichiometry of bound iron following the oxidation reaction, 100 μM DF2 was incubated with excess Fe2+ (3.0 equivalents per dimer using the same procedure as for the kinetic measurements), for 1 h. The resulting solution was chromatographed through a Pharmacia FPLC Peptide column, under aerobic conditions at pH 7.0 (5 mM MOPS/0.15M NaCl, 1.25 mL/min). A single major peak was observed with a retention time corresponding to that expected for a dimer. The concentration of DF2 in the fraction corresponding to the peak was determined by a calibrated reverse phase HPLC assay (Vydak C4 analytical column eluted with a gradient of 9%–72% acetonitrile containing 0.1% TFA), and the concentration of iron was determined using the ferrozine assay (Carter 1971). Guanidine HCl (6 M) was included in the ferrozine assay to denature DF2, because the standard trichloroacetic acid precipitation step did not provide reproducible results with this protein. The extinction coefficient at 300 nm was calculated by subtracting the spectrum of the apo from the diiron forms of the protein.
Acknowledgments
We thank Joseph Jarrett for the use of his glovebox and Natalia Ugulava for assistance with the anaerobic experiment. This work was supported by a grant from the National Institutes of Health (NIH), GM54616, and also partially funded by the MRSEC program of the National Science Foundation (NSF), award DMR9632598.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.52101.
References
- Aberg, A., Nordlund, P., and Ecklund, H. 1993. Unusual clustering of carboxyl side chains in the core of iron-free ribonucleotide reductase. Nature 361 276–278. [DOI] [PubMed] [Google Scholar]
- Andersson, K.K. and Gräslund, A. 1995. Diiron-oxygen proteins. Adv. Inorg. Chem. 43 359–403. [Google Scholar]
- Andersson, M.E., Högbom, M., Rinaldo-Matthis, A., Andersson, K.K., Sjöberg, B.M., and Nordlund, P. 1999. The crystal structure of an azide complex of the diferrous R2 subunit of ribonucleotide reductase displays a novel carboxylate shift with important mechanistic implications for diiron-catalyzed oxygen activation. J. Am. Chem. Soc. 121 2346–2352. [Google Scholar]
- Benson, D.E., Wisz, M.S., Liu, W., and Hellinga, H.W. 1998. Construction of a novel redox protein by rational design: Conversion of a disulfide bridge into a mononuclear iron–sulfur center. Biochemistry 37 7070–7076. [DOI] [PubMed] [Google Scholar]
- Benson, D.E., Wisz, M.S., and Hellinga, H.W. 2000. Rational design of nascent metalloenzymes. Proc. Natl. Acad. Sci. USA 97 6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini, I. and Luchinat, C. 1984. High spin cobalt(II) as a probe for the investigation of metalloproteins. Adv. Inorg. Biochem. 6 71–111. [PubMed] [Google Scholar]
- Bollinger, J.J., Edmondson, D.E., Huynh, B.H., Filley, H., Norton, J.R., and Stubbe, J. 1991. Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science 253 292–298. [DOI] [PubMed] [Google Scholar]
- Brittain, I.J., Huang, X., and Long, E.C. 1998. Selective recognition and cleavage of RNA loop structures by Ni(II) Xaa–Gly–His metallopeptides. Biochemistry 37 12113–12120. [DOI] [PubMed] [Google Scholar]
- Carter, P. 1971. Spectrophotometric determination of serum iron at the submicrogram level with a new reagent (Ferrozine). Anal. Biochem. 40 150–158. [DOI] [PubMed] [Google Scholar]
- Choma, C.T., Lear, J.D., Nelson, M.J., Dutton, P.L., Robertson, D.E., and DeGrado, W.F. 1994. Design of a heme-binding four-helix bundle. J. Am. Chem. Soc. 116 856–865. [Google Scholar]
- da Silva, J.J.R.F. and Williams, R.J.P. 1993. The biological chemistry of the elements. Oxford University Press, New York, NY.
- Dillon, P.J. and Rosen, C.A. 1993. Use of the polymerase chain reaction for the rapid construction of synthetic genes. PCR Protocols: Current Methods and Applications 15 263–268. [DOI] [PubMed] [Google Scholar]
- Feig, A.L. and Lippard, S.J. 1994. Reactions of non-heme iron(II) centers with dioxygen in biology and chemistry. Chem Rev. 94 759–805. [Google Scholar]
- Fox, B.G., Shanklin, J., Ai, J., Loehr, T.M., and Sanders-Loehr, J. 1994. Resonance Raman evidence for an Fe–O–Fe center in stearoyl-ACP desaturase. Primary sequence identity with other diiron-oxo proteins. Biochemistry 33 12776–12786. [DOI] [PubMed] [Google Scholar]
- Frolow, F., Kalb A.J., and Yariv, J. 1994. Structure of a unique twofold symmetric haem-binding site. Nature Struc. Biol. 1 453–460. [DOI] [PubMed] [Google Scholar]
- Furst, A. 1997. The XL-I analytical ultracentrifuge with Rayleigh interference optiics. Eur. Biophys. J. 35 307–310. [Google Scholar]
- Gibney, B.R., Mulholland, S.E., Rabanal, F., and Dutton, P.L. 1996. Ferredoxin and ferredoxin-heme maquettes. Proc. Natl. Acad. Sci. USA 93 15041–15046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel, T. and DeGrado, W.F. 1990. De novo design of a Zn2+-binding protein. J. Am. Chem. Soc. 112 6710–6711. [Google Scholar]
- Handel, T.M., Williams, S.A., and DeGrado, W.F. 1993. Metal ion-dependent modulation of the dynamics of a designed protein. Science 261 879–885. [DOI] [PubMed] [Google Scholar]
- Hay, R.W., Hassan, M.M., and You-Quan, C. 1993. Kinetic and thermodynamic studies of the copper (II) and nickel (II) complexes of Glycylglycyl-l-histidine. J. Inorg. Biochem. 52 17–25. [DOI] [PubMed] [Google Scholar]
- Hellinga, H.W. 1998. The construction of metal centers in proteins by rational design. Folding and Design 3 R1–R8. [DOI] [PubMed] [Google Scholar]
- Huheey, J.E., Keiter, E.A., and Keiter, R.L. 1993. Inorganic chemistry: Principles of structure and reactivity. Harper Collins College Publishers, New York, NY.
- Keech, A.M., Le Brun, N.E., Wilson, M.T., Andrews, S.C., Moore, G.R., and Thomson, A.J. 1997. Spectroscopic studies of cobalt(II) binding to Escherichia coli bacterioferritin. J. Biol. Chem. 272 422–429. [DOI] [PubMed] [Google Scholar]
- Kharakoz, D.P. 1997. Partial volumes and compressibilities of extended polypeptide chains in aqueous solution: Addititivity scheme and implication of protein unfolding at normal and high pressure. Biochemistry 36 10276–10285. [DOI] [PubMed] [Google Scholar]
- Lange, S.J. and Que, L., Jr. 1998. Oxygen activating nonheme iron enzymes. Curr. Opin. Chem. Biol. 2 159–172. [DOI] [PubMed] [Google Scholar]
- Laue, T., Shaw, B.D., Ridgeway, T.M., and Pelletier, S.L. 1992. Analytical ultracentrifugation in biochemistry and polymer science. The Royal Society of Chemistry, Cambridge, UK, pp. 90–125.
- Le Brun, N.E., Keech, A.M., Mauk, M.R., Mauk, A.G., Andrews, S.C., Thomson, A.J., and Moore, G.R. 1996. Charge compensated binding of divalent metals to bacterioferritin: H+ release associated with cobalt(II) and zinc(II) binding at dinuclear metal sites. FEBS Lett. 397 159–163. [DOI] [PubMed] [Google Scholar]
- Lindqvist, Y., Huang, W., Schneider, G., and Shanklin, J. 1996. Crystal structure of Δ9 stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other di-iron proteins. EMBO J. 15 4081–492. [PMC free article] [PubMed] [Google Scholar]
- Lombardi, A., Summa, C.M., Geremia, S., Randaccio, L., Pavone, V., and DeGrado, W.F. 2000. Retrostructural analysis of metalloproteins: Application to the design of a minimal model for diiron proteins. Proc. Natl. Acad. Sci. USA 97 6298–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund, P. and Eklund, H. 1995. Di-iron-carboxylate proteins. Curr. Opin. Struct. Biol. 5 758–766. [DOI] [PubMed] [Google Scholar]
- Pessi, A., Bianchi, E., Crameri, S., Venturini, S., Tramontano, A., and Sollazzo, M. 1993. A designed metal-binding protein with a novel fold. Nature 362 367–369. [DOI] [PubMed] [Google Scholar]
- Pinto, A., Hellinga, H.W., and Caradonna, J.P. 1997. Construction of a catalytically active iron superoxide dismutase by rational design. Proc. Natl. Acad. Sci. USA 94 5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabanal, F., DeGrado, W.F., and Dutton, P.L. 1996. Toward the synthesis of a photosynthetic reaction center maquette: A cofacial porphyrin pair assembled between two subunits of a synthetic four-helix bundle multiheme protein. J. Am. Chem. Soc. 118 473–474. [Google Scholar]
- Regan, L. 1995. Protein design: Novel metal-binding sites. Trends Biochem. Sci. 20 280–285. [DOI] [PubMed] [Google Scholar]
- Regan, L. and Clarke, N.D. 1990. A tetrahedal zinc(II)-binding site introduced into a designed protein. Biochemistry 29 10878–10883. [DOI] [PubMed] [Google Scholar]
- Robertson, D.E., Farid, R.S., Moser, C.C., Urbauer, J.L., Mulholland, S.E., Pidikiti, R., Lear, J.D., Wand, A.J., DeGrado, W.F., and Dutton, P.L. 1994. Design and synthesis of multi-haem proteins. Nature 368 425–432. [DOI] [PubMed] [Google Scholar]
- Schellman, C. 1980. The αL-conformation at the ends of helices. In Protein Folding (ed. R. Jaenicke), pp. 53–61. Elsevier, North-Holland, NY.
- Stemmler, A.J., Burrows, C.J. 1999. The Sal-XH motif for metal-mediated oxidative DNA–peptide cross-linking. J. Am. Chem. Soc. 121 6956–6957. [Google Scholar]
- Tolman, W.B., Liu, S., Bentsen, J.G., and Lippard, S.J. 1991. Models of the reduced forms of polyiron-oxo proteins: An asymmetric, triply carboxylate bridged diiron(II) complex and its reaction with dioxygen. J. Am. Chem. Soc. 113 152–164. [Google Scholar]
- Torrado, A., Walkup, G.K., and Imperiali, B. 1998. Exploiting polypeptide motifs for the design of selective Cu(II) ion chemosensors. J. Am. Chem. Soc. 120 609–610. [Google Scholar]
- Vriend, G. 1990. A molecular modeling and drug design program. J. Mol. Graph. 8 52–56. [DOI] [PubMed] [Google Scholar]
- Waller, B.J. and Lipscomb, J.D. 1996. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem. Rev. 96 2625–2657. [DOI] [PubMed] [Google Scholar]
- Yang, X., Chen-Barrett, Y., Arosio, P., and Chasteen, N.D. 1998. Reaction paths of iron oxidation and hydrolysis in horse spleen and recombinant human ferritins. Biochemistry 37 9743–9750. [DOI] [PubMed] [Google Scholar]
- Yang, X., Le Brun, N.E., Thomson, A.J., Moore, G.R., and Chasteen, N.D. 2000. The iron oxidation and hydrolysis chemistry of Escherichia coli bacterioferritin. Biochemistry 39 491–523. [DOI] [PubMed] [Google Scholar]