

Abstract
A function annotation method using the sequence-to-structure-to-function paradigm is applied to the identification of all disulfide oxidoreductases in the Saccharomyces cerevisiae genome. The method identifies 27 sequences as potential disulfide oxidoreductases. All previously known thioredoxins, glutaredoxins, and disulfide isomerases are correctly identified. Three of the 27 predictions are probable false-positives. Three novel predictions, which subsequently have been experimentally validated, are presented. Two additional novel predictions suggest a disulfide oxidoreductase regulatory mechanism for two subunits (OST3 and OST6) of the yeast oligosaccharyltransferase complex. Based on homology, this prediction can be extended to a potential tumor suppressor gene, N33, in humans, whose biochemical function was not previously known. Attempts to obtain a folded, active N33 construct to test the prediction were unsuccessful. The results show that structure prediction coupled with biochemically relevant structural motifs is a powerful method for the function annotation of genome sequences and can provide more detailed, robust predictions than function prediction methods that rely on sequence comparison alone.
Keywords: Disulfide oxidoreductase, fuzzy functional forms (FFFs), protein function prediction, oligosaccharyltransferase (OST), OST3, OST6, N33, structural genomics
The success of the large-scale genome sequencing projects has provided a deluge of sequence information. For this information to be used, the biological significance of each sequence must be established. Thus, methods have been developed for identifying biological functions from sequence information, with most depending on an evolutionary relationship between sequences.
Because structure is more conserved than sequence, structural information should enhance protein function prediction (Skolnick et al. 2000). However, the structure of a protein alone is insufficient for accurate function prediction (Hegyi and Gerstein 1999). To address this problem, we recently developed a method that uses three-dimensional structural information to identify biologically relevant sites in protein structures; the resulting active site descriptors are termed fuzzy functional forms (FFFs) (Fetrow and Skolnick 1998). This method has been shown to be specific both for high-resolution experimental structures as well as inexact protein models produced by threading and ab initio modeling methods and has identified proteins likely to show the disulfide oxidoreductase and α/β hydrolase functions in proteins from the Escherichia coli genome (Fetrow et al. 1998; Zhang et al. 1998). We note that alternative approaches that have been developed to describe active sites need high-resolution models to work (Wallace et al. 1997).
Here, we expand disulfide oxidoreductase function analysis of the sequences of the yeast, Saccharomyces cerevisiae. We show that structural information followed by FFF filtration can validate predictions made by other methods, can make predictions about sequences unrecognized by sequence-only methods, and can identify specific functional residues in a protein. A detailed comparison to the sequence motif method, Blocks (Pietrovski et al. 1996), shows the higher specificity of the structure-based FFF approach. The results show that automated structural analysis coupled with specific biochemical information is a very powerful approach for functional annotation.
Results
Disulfide oxidoreductase FFF, a structural motif for function identification
The FFF for the disulfide oxidoreductase active site found in the glutaredoxins, thioredoxins, and disulfide isomerase proteins is based on the common active site of this diverse protein family. Two cysteines, located at the N terminus of an α helix, are essential for the redox activity (Yang and Wells 1991a, 1991b; Kortemme and Creighton 1995; Dyson et al. 1997). A structurally conserved cis-proline is located in close spatial proximity but far in sequence to the cysteines (Chaohong et al. 1997). The disulfide oxidoreductase FFF consists of the distances between alpha carbons with a small variance (Fetrow and Skolnick 1998).
FFF analysis of sequences in the S. cerevisiae genome
The S. cerevisiae genome was screened for proteins that might show disulfide oxidoreductase function. Its 6215 open reading frames (ORFs) were threaded onto a nonredundant structural database (see Materials and Methods), and the alignments were analyzed by the FFF. The 27 sequences predicted to show disulfide oxidoreductase activity by our automated sequence/structural/functional analysis are shown in Table 1.
Table 1.
Proteins in Saccharomyces cerevisiae genome predicted to have the thiol-disulfide oxidoreductase active site
| Functional motifb | Active site | ||||||||
| Sequence IDa | Thd/FFFc | CPd | Pr | PS | Bl | C | C/S | P | Database description |
| YLR043C (trx1) | X | X | X | X3e | Xc | 30 | 33 | 73 | Thioredoxin 1 |
| YGR209C (trx2) | X | X | X | X3c | Xe | 31 | 34 | 74 | Thioredoxin 2 |
| YCR083W (trx3) | X | X | X | X5c | Xf | 55 | 58 | 98 | Thioredoxin 3 |
| YDR513W (ttr1) | X | X | X | X4c | X | 61 | 64 | 110 | Glutaredoxin |
| YCL043C (PDI1) | X | X | — | X8c | Xc | 61 | 64 | 106 | PDI |
| X | X | X | X8c | Xc | 405 | 409 | 451 | ||
| YCL035C (YCD5) | X | X | X | X3c | X | 27 | 30 | 76 | Possible glutaredoxin |
| YCR288C (MPD1) | X | X | X | X10c | Xe | 59 | 62 | 105 | PDI related |
| YIL005W | X | X | — | X8f | Xc | 60 | 63 | 109 | Putative PDI |
| — | ? | X | 60 | 63 | 409 | ||||
| YLR364W | X | X | X2 | — | X2e | 25 | 28 | 74 | Hypothetical glutaredoxin-like |
| YBR014C | X | X | X | — | X | 108 | 111B | 156 | Possible glutaredoxin |
| YDL010W | X | X | — | — | X | 136 | 139B | 184 | Hypothetical protein |
| YDR518W (EUG1) | X | X | — | — | X | 62 | 65B | 107 | Possible disulfide isomerase |
| — | X | X | — | Xe | 405 | 408B | 453/459 | ||
| YOL088C (MPD2) | X | — | — | — | Xe | 56 | 59 | 96 | Possible disulfide isomerase |
| X | X | X | — | Xe | 56 | 59 | 106 | ||
| YER174C | X | X | X? | — | X3? | 171 | 174B | 213 | Hypothetical protein (now grx4) |
| YDR098C | X | X | X? | — | X4? | 211 | 214B | 253 | Possible thioredoxin (now grx3) |
| YPL059W | X | X | — | — | X2f | 60 | 63B | 105 | Possible glutared (now grx5) |
| YOR085W (OST3) | X | X | X2 | — | — | 73 | 76 | 133 | Oligosaccharyltransferase |
| YML019W (OST6) | X | X | — | — | — | 78 | 81 | 127 | Possible oligosaccharyltransferase |
| YDR286C | X | ? | — | — | X2e | 31 | 34 | 79 | Hypothetical protein |
| YNL155W | — | ? | X8e | — | — | 18 | 21 | ? | Hypothetical protein |
| X | ? | — | — | — | 31 | 34 | 164 | ||
| YDR133C | X | ? | — | — | — | 74 | 75 | 94 | Hypothetical protein |
| YDR199W | X | ? | — | — | — | 37 | 40 | 59 | Hypothetical protein |
| YOL024W | X | ? | — | — | — | 30 | 43B | 149 | Hypothetical protein |
| YKL102C | X | ? | — | — | — | 28 | 48 | 82 | Hypothetical protein |
| YLR245C (cdd1) | X | — | — | — | — | 59 | 61 | 112 | Cytidine deaminase |
| — | — | X5 | — | — | 96 | 99 | 112 | ||
| YLR246W (erf2) | X | — | — | — | — | 175 | 178 | 297 | Hypothetical protein |
| YHR002W | X | — | — | — | — | 198 | 201 | 299 | Possible mitochondrial carrier protein |
Question marks indicate that the sequence was identified as a thioredoxin by the Prints or Blocks motif libraries, but instead of the CXXC motif, an AXXC motif was found. For sequence YER174C the active site identified by the motif library was found to be A34 C36 P158, and for sequence YDR098C it was found to be A69 C72 P197. The same site, containing AXXC, was identified by both Prints and Blocks in each protein. The FFF identifies as alterative active site, containing the appropriate CXXC motif.
a Sequence number in the Saccharomyces genome sequence database.
b Search of each sequence found by Thread/FFF protocols against the local signature database (Prints (Pr; http://www.biochem.ucl.ac.uk/cgibin/attwood/SearchPrintsForm2.pl), Prosite (PS; http://expasy.hcuge.ch/sprot/acnpsit1.html), or BLocks (Bl; http://www.blocks.fhcrc.org/blocks_search.html). The number following the X indicates the rank of the sequence in the search.
c Top six alignments of S. cerevisiae operating from ORF to 1bed, 1ego, 1erv, 1fvk (chain A), 1kte, 1thx, 1tof, or 2trx (chain A), using threading (Jaroszewski et al. 1998), followed by scanning the sequence–sequence alignment for the active site residues specified by the FFF for the thiol-disulfide oxidoreductase activity of the glutaredoxin/thioredoxin family (Fetrow and Skolnick 1998).
d Conservation profile of homologous sequences demonstrates whether conservation of all three active site residues is >50%. For those sequences marked by a question mark, too few homologous sequences were found, so construction of a conservation profile was not possible.
e P motif not found.
f CXXC motif not found.
(FFF) fuzzy functional form; (CP) conservation profile; (Pr) Prints; (PS) Prosite; (Bl) Blocks; (C) cysteine; (C/S) cysteine or serine; (P) proline; (PDI) protein disulfide isomerase.
Of these 27 sequences, 18 predictions are supported by the conservation profile analysis (Table 1), for which we calculate the conservation of predicted active site residues in homologous sequences (Zhang et al. 1998). Of these 18, three are known thioredoxins (YLR043C, also known as trx1; YGR209C or trx2; and YCR083W or trx3; Muller 1992; Pedrajas et al. 1999), a known glutaredoxin (YDR513W or ttr1), and a known disulfide isomerase (YCL043C or PDI1; Farquhar et al. 1991; Tachikawa et al. 1991). Eleven are annotated in the genome database as possible glutaredoxins, thioredoxins, protein disulfide isomerases (PDIs), or hypothetical proteins. For those sequences identified by the FFF and validated by the conservation profile, the disulfide oxidoreductase functional annotation is presented as further support for sequence-based function predictions or, when the function was not previously annotated, as novel predictions.
Six of the 27 FFF predictions cannot be confirmed (marked with a question mark in the CP column in Table 1), because too few proteins are homologous with these sequences to calculate the conservation profile. All six are hypothetical proteins; thus, they are presented as predictions.
For an additional three sequences, YLR245C, YLR246W, and YHR002W, the conservation profile does not support the FFF prediction indicating that the proposed functional residues are not conserved. There are two possible reasons for this. First, the functional residue predictions might be incorrect, and the conservation profile helps identify false-positives. Second, the protein family might be composed of subfamilies, in which the predicted functional residues are only conserved in one of the subfamilies. For example, subfamilies within a larger sequence superfamily, such as the serine-threonine phosphatases, can produce inaccurate conservation profile results, if the subfamilies are not distinguished (Fetrow et al. 1999). Thus, we performed a cluster analysis of GenBank sequences that aligned to YLR245C, YLR246W, and YHR002W, respectively. The CXXC and proline residues were not conserved, suggesting that these three sequences are false-positives.
Analysis of the predictions and general comparison to the sequence motif libraries
The 27 sequences found by the FFF were analyzed by the Prints, Prosite, and Blocks sequence motif databases with the results shown in Table 1. Prosite contains two motifs, one for the glutaredoxins (PDOC00173) and one for the thioredoxins (PDOC00172). Each encompasses only the CXXC motif. BL00194 is the only thioredoxin block described in the Blocks database and encompasses the CXXC sequence motif. Glutaredoxins are identified by two blocks, BL00195A and BL00195B. BL00195A includes the CXXC motif, and BL00195B includes the proline motif. Prints uses three sequence motifs for the glutaredoxin and thioredoxin families: one encompasses the CXXC motif, the second the proline motif, and the third is a sequence region not used by the FFF.
Eight yeast sequences are identified as disulfide oxidoreductases by Prints, Prosite, and Blocks (Table 1). All are either well studied or trivially homologous with known disulfide oxidoreductases; we term these consensus positives, to distinguish them from the experimentally known true-positives. All consensus positives in the yeast genome are hit by the FFF.
One of the eight, YCL043C or PDI1, is predicted to show two different disulfide oxidoreductase domains or distinct active sites by the FFF (Table 1). Both thioredoxin-like sites were suggested when the PDI1 gene originally was isolated (Farquhar et al. 1991; Tachikawa et al. 1991), and both are experimentally validated, although there is some debate about which is the more active (Holst et al. 1997; Westphal et al. 1999). Both active sites are identified by the FFF, Prosite, and Blocks and validated by the conservation profile. However, the Web-based version of Prints identifies only the active site of the second domain. In the first domain, Prosite and Blocks only identified the CXXC motif of both domains. Thus, the complete active site, including both the CXXC and the proline were only identified in both domains by the FFF, and not by the sequence motif methods alone.
YIL005W is another consensus positive, because it is identified as a disulfide oxidoreductase by all methods (Table 1). However, the exact active site proline is again not clearly identified by the sequence motif methods alone, whereas the same cysteines, Cys60 and Cys63, are. The Prosite and Blocks thioredoxin motifs only use one sequence motif that contains the active site CXXC, so the proline is not explicitly identified by these motifs. Prints does identify a proline, Pro409; however, the conservation profile cannot be validated because there are too few homologous sequences with this domain. The FFF identifies a different active site proline, Pro109, and the conservation profile analysis validates this proline (Table 1). YIL005W is annotated as a putative PDI, and we propose that the active site residues are Cys60, Cys63, and Pro109, as uniquely identified by the FFF. Thus, for eight consensus positive sequences found in the yeast genome, the FFF aids in the identification of the complete active site in two cases.
The FFF also helps confirm several predictions made by only one or two of the sequence motif methods, including YLR364W, YBR014C, YDL010W, YDR518W, and YOL088C (Table 1). YLR364W is annotated as a hypothetical glutaredoxin-like protein. It is identified as a glutaredoxin by Prints and Blocks as the second highest hit by both methods. The FFF/conservation analysis predicts this is a glutaredoxin with specified putative active site residues.
Three sequences, YBR014C, YDL010W, and YDR518W, are predicted as disulfide oxidoreductases with a serine instead of a second cysteine. Disulfide oxidoreductases can still perform limited functions, including disulfide exchange, when the second cysteine is a serine (Bushweller et al. 1992; Yang et al. 1998); thus, the prediction is reasonable. YBR014C is annotated as a glutaredoxin homolog, YDL010W a hypothetical protein, and YDR518W (EUG1) as a possible PDI (Table 1). Experimentally, overexpression of EUG1 complements a PDI1 deletion in yeast (Tachibana and Stevens 1992), providing experimental evidence for the FFF prediction. The other two predictions provide additional confirmation of the weak predictions made by the sequence motif libraries. Again the FFF protocol provided identifies the putative active site residues.
Finally, the sequence YOL088C (MPD2) is predicted to be a disulfide oxidoreductase by Prints and Blocks, but not by Prosite and is consistent with the FFF methodology (Table 1). Overexpression of MPD2 is known to complement a deletion of the PDI1 gene (Tachikawa et al. 1997), providing experimental evidence in support of the prediction. The FFF predicts two sites for YOL088C: the same two cysteines are identified, but different proline residues are identified by different scoring functions in the threading algorithm (see Materials and Methods). The conservation profile analysis shows that only one of the identified prolines is conserved in homologous sequences, suggesting that this protein will only contain one true disulfide oxidoreductase active site.
Two function predictions show some of the pitfalls of the scoring methods used in Prints and Blocks and exemplify the advantages of the FFF method. YER174C and YDR098C are identified as glutaredoxins or thioredoxins by both Prints and Blocks; however, the active site identified by both methods contains an AXXC motif, rather than a CXXC motif (Table 1). Mutagenesis experiments strongly suggest that when a serine replaces the first cysteine, the protein can no longer perform disulfide oxidoreductase chemistry (Walker et al. 1996). Given these data, it is unlikely that the AXXC motif, in which alanine replaces the first cysteine, would be a functional disulfide oxidoreductase. The FFF analysis identifies a different possible active site motif in each of these sequences that both contain the CXXS motif and both are validated by the conservation profile analysis. Some glutaredoxins with limited activity are known to contain a serine at the position of the second cysteine (Bushweller et al. 1992; Yang et al. 1998); thus, we suggest that the FFF has identified active sites in these proteins that are more likely to be correct than those identified by Prints and Blocks.
Novel predictions, subsequently validated
Subsequent to our initial analysis, but before our submission, we learned that these two novel FFF predictions have been experimentally validated. According to the Saccharomyces Genome Database (SGD; http://genome-www.stanford.edu/Saccharomyces), YER174C is glutaredoxin 4, responsible for protecting yeast from oxidative damage (Rodriguez et al. as referenced in http://genome-www.stanford.edu/Saccharomyces). In addition, YDR098C is annotated as glutaredoxin 3 (http://genome/www.stanford.edu/Saccharomyces).
The FFF analysis also identifies YPL059W as a disulfide oxidoreductase (Table 1). Web-based versions of Prints and Prosite did not identify this sequence as a glutaredoxin or a thioredoxin. Web-based Blocks only identified it as the second highest hit; furthermore, Blocks only identified the proline motif, not the required active site cysteines. Thus, a manual, knowledge-based analysis would disregard this Block result because the essential active site cysteines were not identified. Using the FFF, we automatically identified a CXXS motif that satisfies the structural requirements of a glutaredoxin active site. Thus, a complete active site was automatically identified only by the FFF methodology, not by any of the sequence motif libraries. Subsequently, according to the SGD Web site, this sequence was shown experimentally to be a yeast glutaredoxin and was named glutaredoxin 5 and is responsible for protection of yeast against oxidative damage (Rodriguez et al. as referenced in http://genome-www.stanford.edu/Saccharomyces). When published, these data will experimentally validate this novel prediction made by the FFF approach.
A novel prediction for function in the N-oligosaccharyltransferase subunits
The FFF methodology makes another novel prediction for two sequences that are not clearly made by any other method. The FFF predicts YOR085W (OST3) and YML019W (OST6) as disulfide oxidoreductases and both are validated by the conservation profile (Table 1). The proposed structures with the putative active sites are shown in Figure 1 ▶. The prediction for YML019W could not be made clearly by either the sequence motif methods or by standard sequence alignment methods, although Prints does identify OST3 as a glutaredoxin as the second highest scoring hit. OST3 is the 34-kD γ subunit of the oligosaccharyltransferase complex in yeast (Karaoglu et al. 1995). OST6 is the recently described subunit of the same complex with similar membrane topology to OST3 (Knauer and Lehle 1999a,b). Either Δost3 or Δost6 mutants cause only minor defects in N-glycosylation; however, the double Δost3Δost6 mutants yield severely underglycosylated membrane-bound and soluble proteins, suggesting that these subunits have some overlapping activity (Knauer and Lehle 1999a,b). The OST complex isolated from Δost3Δost6 strain shows reduced activity in vitro (Knauer and Lehle 1999a,b). Apparently, one or the other of OST3 and OST6 is needed for optimal oligosaccharyl transfer, possibly through some regulatory activity or in positioning the complex for different translocation pathways (Knauer and Lehle 1999a,b). From the results presented in Table 1 and the models presented in Figure 1 ▶, we propose that the overlapping activity found in both OST3 and OST6 is an oxidoreductase activity localized to the extramembrane domain. The redox activity could be regulatory, or could be important for properly positioning the various elements of the complex. Our analysis cannot distinguish these possibilities; however, the prediction of a specific disulfide oxidoreductase site in these proteins provides a hypothesis that now can be tested experimentally.
Fig. 1.

α-Carbon traces of the three-dimensional structural models of the N33 gene product from human (A) and the OST3 gene product from the S. cerevisiae genome (B). (Black circles) Putative disulfide oxidoreductase active site residues that were identified by the FFF. Models were produced from the threading alignment.
Prediction of the function of a homologous gene from Caenorhabditis elegans and the N33 gene product from human
OST3 is homologous with several other protein sequences found in GenBank, namely an ORF of unknown function from C. elegans and an ORF from the N33 region of the human chromosome (MacGrogan et al. 1996; Knauer and Lehle 1999a,b). The sequence identity between OST3 and the C. elegans protein is 43%, whereas that between OST3 and the N33 ORF is ∼20% (Knauer and Lehle 1999a,b). But, based on hydrophobicity calculations, all show strikingly similar membrane topologies, and it has been suggested that they have similar functions.
N33 is found on a section of human chromosome band 8p22 (MacGrogan et al. 1996). Homozygous deletion is highly correlated with metastatic prostate cancer (MacGrogan et al. 1996). Allelic deletion is also associated with colorectal and pancreatic cancers (Levy et al. 1999). Such observations suggest that N33 is a cell-specific tumor suppressor gene, and the researchers suggested a subtle regulatory activity for this gene product (MacGrogan et al. 1996; Levy et al. 1999). The relationship between the N33 ORF and the OST subunits is unknown, but OST3 and OST6 may have some overlapping regulatory activity, and that loss of this activity may contribute to carcinogenesis (Knauer and Lehle 1999a,b).
Because of the similarity between OST3, OST6, and N33 previously noted in the literature, we independently ran the N33 ORF protein sequence through our threading/FFF protocol and found that its extramembrane domain also is predicted to have a disulfide oxidoreductase active site, a novel biochemical function prediction for this protein. We suggest that the specific activity of N33, lost in some forms of metastatic prostate cancer and other cancers, is oxidoreductase activity or a redox regulatory mechanism.
Experimental verification of oxidoreductase activity in the predicted sequence of N33 was attempted by synthesizing the gene, followed by overexpression of the protein in E. coli. The protein was expressed into inclusion bodies in the bacterial expression system, and it proved extremely difficult to solubilize and purify the protein, and subsequently to fold it into a globular domain. Assays for oxidoreductase activity (Holmgren and Bjornstedt 1995) showed only low activity that could not be distinguished unequivocally from background. Mutants were constructed in which one or more of the putative active site cysteine residues were changed to alanine. The mutant proteins were subjected to the same assay conditions and gave similar results to the wild-type protein. On this basis, it could be concluded that the present N33 construct was not active as a specific thiol oxidoreductase; however, an alternative explanation is that the N33 construct was unfolded or not properly folded.
To test this idea, expressed protein was subjected to both circular dichroism (CD) and nuclear magnetic resonance (NMR) spectroscopy. The results for the two constructs are shown in Figure 2 ▶. The CD spectrum of the shorter construct, residues 61–180, shows little evidence for regular secondary structure (Fig. 2A ▶). Its NMR spectrum has quite narrow line widths at pH 3.4 (Fig. 2C ▶, black cross peaks), but the 1H resonance dispersion is very small, indicating that it is unfolded and monomeric. The loss of cross peak signals in the NMR spectrum at pH 5.0 (Fig. 2C ▶, red cross peaks) is most likely an indication that the resonances are broadened beyond detection by aggregation. The few observed resonances are broad and are also located in the center of the spectrum, indicating that these residues are unfolded even in the aggregated protein. The results for the longer construct, residues 61–201, show the presence of more secondary structure at higher pH in the CD spectrum (Fig. 2B ▶). This construct is also unfolded and apparently monomeric at pH 3.6 (Fig. 2D ▶, black cross peaks). Resonance dispersion at pH 6.0 (Fig. 2D ▶, red cross peaks) indicates that the protein is folded, but the broadness of the resonance line widths (with consequent loss of signal) is again indicative of a high molecular weight, most likely due to aggregation of the folded protein in this case. Reduction of the protein concentration, which can alleviate concentration-dependent aggregation, is not an option, because the protein concentration (∼50–100 μM) is already as low as it can be for NMR.
Fig. 2.

Summary of experimental data on two constructs of N33. (A) Circular dichroism (CD) spectra of the shorter construct, 61–180 at two pH values, 6.0 and 3.0. (B) CD spectra of the longer construct (61–201) at two pH values, 6.0 and 3.7. (C) 1H-15N HSQC spectrum (500 MHz) of 50 μM 61–180 at pH 3.4 (black data points) and at pH 5.4 (red data points). Lower numbers of cross peaks are observed at the higher pH, probably because of aggregation of the protein. (D) 1H-15N HSQC spectrum (500 MHz) of 95 μM 61–201 at pH 3.6 (black data points) and at pH 6.0 (red data points). The protein at the higher pH appears to be folding, but the signal is extremely weak, an indication that much of it probably is aggregated.
Although the immediate inference from these data is that the hypothesis concerning the activity of N33 is incorrect, the lack of activity could also be due to several factors, including a wrongly folded or unfolded structure (as suggested by the CD or NMR data), or that achieving a stable, correct fold is dependent on the presence of a partner protein. By analogy, OST3, the yeast N33 homolog, interacts strongly with Stt3P, another subunit of the yeast oligosaccharyltransferase enzyme complex (Karaoglu et al. 1997; Knauer and Lehle 1999a,b). Thus, proper folding of N33 could require a partner protein, analogous to the yeast Stt3P protein.
Is the threading score alone enough to predict protein function?
The above results of prediction of disulfide oxidoreductases in the yeast genome suggest that the combination of threading and FFF analysis produces higher quality functional annotations than the sequence motif–based methods. Because we use a threading-type sequence-to-structure alignment, the obvious next question is: is the threading significance score itself enough to predict the protein function? Given the plethora of proteins with similar structures and dissimilar functions, one would guess that the answer would be no; however, we tested it directly. A histogram of the threading scores shows that threading scores for sequences that are unlikely to show disulfide oxidoreductase function are intermixed with the scores of sequences that are likely to show the function (Fig. 3 ▶). These graphs provide direct evidence that threading score alone, although adequate for some level of structure prediction, is insufficient for protein function prediction in general. Additional biochemical or functional information must be included to validate the threading models, as originally suggested by Lathrop and Smith (1996). FFF analysis is an automated method for doing so.
Fig. 3.
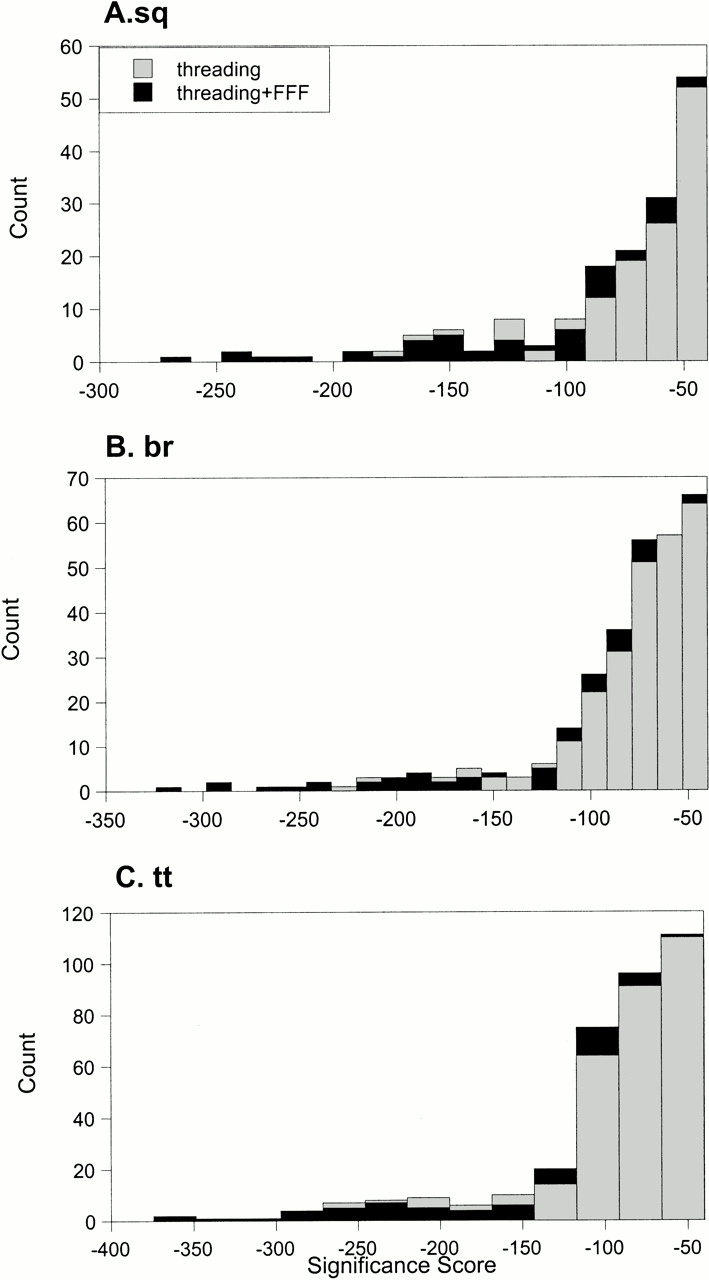
Distribution of threading significance scores for the S. cerevisiae genome sequences. More significant scores are more negative. The three plots are distributions of different threading scoring functions: (A) sq, or sequence-sequence method; (B) br, or sequence-structure method; and (C) tt, structure-predicted structure method. These scoring methods are detailed in Jaroszewski et al. (1998). (Black bars) Threading scores for sequences in which a disulfide oxidoreductase active site was identified in the threading model. (FFF) fuzzy functional form.
Genome analysis using Blocks, a sequence motif database
The comparison of the FFF analysis to the sequence motif database results presented in Table 1 is misleading. One FFF structural motif was compared with many sequences, whereas only one sequence was compared with a library of sequence motifs. This one-to-many comparison for the FFF methodology versus the many-to-one comparison for the motif libraries does not allow a comparable evaluation of the two methods. To more directly compare the performance of the sequence motif databases against the FFF structural motifs, we selected two Blocks motifs and scored them against all sequences in the S. cerevisiae genome. Blocks was chosen because it identifies more sequences than either Prints or Prosite (Table 1; unpublished results). As described above, BL00194 is the thioredoxin block. It encompasses the CXXC active site sequence motif. Glutaredoxins are identified by two blocks, BL00195A and BL00195B. BL00195A includes the CXXC active site motif, whereas BL00195B encompasses the proline motif.
The results of the search of all sequences in the S. cerevisiae genome with the glutaredoxin and thioredoxin Blocks are shown in Figure 4 ▶. For the thioredoxin block, eight sequences with scores above 870 are consensus positives. These sequences are identified by the Web-based version of Blocks and the FFF and are validated further by the conservation profile analysis of the FFF-predicted functional residues. However, the next sequence, with a Blocks score of ∼850, does not appear to be a disulfide oxidoreductase. The N terminus of this protein has weak similarity to several transcriptional regulatory proteins. The sequence is not identified as a disulfide oxidoreductase by either the FFF or the Web-based versions of the other motif libraries. This protein has many cysteines, including several CXXC sequences, but its similarity to regulatory proteins and its many cysteines suggests a metal-binding protein, such as a zinc finger. Next follows a sequence, YDR098C, which is predicted to be a disulfide oxidoreductase by the FFF, a prediction that is validated by the conservation of its active site residues (Table 1). Although Blocks correctly identifies this sequence, the active site of this protein is likely incorrectly identified because a different motif, AXXC rather than CXXC, is identified (see Table 1 and discussion above). Five other sequences identified by Web-based Blocks and the FFF and validated by the conservation profile analysis are found with lower Blocks scores, ranging from ∼775 to 820 (Fig. 4A ▶). Twenty sequences that are not identified by any method and are likely to be false-positive sequences are found in this same score range. In summary, if the sequences that are hit by Blocks, the FFF, and the conservation profile are, in this case, considered to be consensus positives, one has to wade through 21 probable false-positives to find all consensus positive thioredoxins in the S. cerevisiae genome, by using the Blocks methodology. Similar application of the FFF methodology identifies only 11 potential false-positives (Table 1).
Fig. 4.
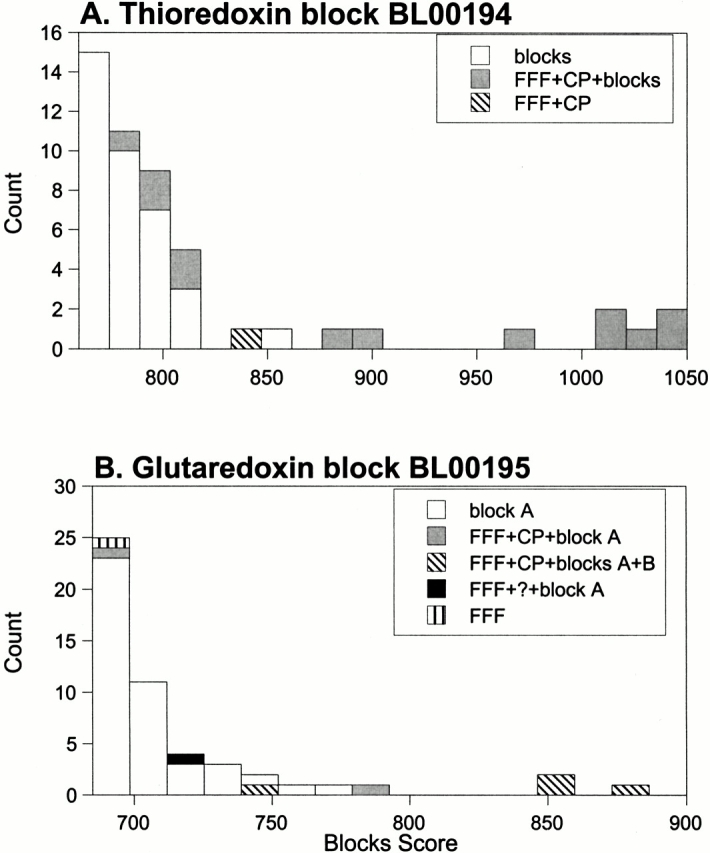
Distributions of Blocks scores for all sequences in the S. cerevisiae genome: (A) thioredoxin block (BL00194) and (B) glutaredoxin blocks (BL00195A and BL00195B). (A) Gray bars represent sequences that were identified by BL00194 and by the FFF and validated by the conservation profile. For BL00194, the gray bars are assumed to be the consensus positives discussed in the text. (Hatched bar) Sequence (YDR098C) identified by the FFF and validated by the conservation profile, but it was not properly identified by the Web-based version of Blocks (see Table 1). (White bars) Remaining sequences identified by BL00194, and most are likely false-positives (see text). (B) Gray bars represent sequences that were identified by BL00195A and the FFF, then validated by the conservation profile. (Diagonal hatched bars) Sequences identified by both BL00195A and B, and the FFF, and validated by the conservation profile. For BL00195, the gray and diagonal hatched bars are assumed to be the consensus positives discussed in the text. (Black bar) Sequence (YDR286C) identified by BL00195A and the FFF. We are unable to validate this prediction by using the conservation profile (see Table 1). (Vertical hatched bar) Sequence (YLR246W) identified by the FFF, but not validated by the conservation profile. This sequence also was not found as one of the top 10 scores by the Web-based version of Blocks (see Table 1). (White bars) The remaining sequences identified by BL00195A, and most are likely false-positives (see text).
A similar story can be seen when the yeast genome is analyzed by the glutaredoxin blocks (Fig. 4B ▶). Here, one would have to wade through 43 potential false-positives, including a hypothetical endopeptidase and a putative Zn finger to identify all consensus positive glutaredoxins in the genome.
Discussion
The value of structural information in protein function prediction
The ultimate goal of the large-scale genome projects is to understand all levels of function, including biochemical function, cellular function, metabolic function, and phenotypic function, of all the gene products. Most often, such functional annotation is accomplished via sequence similarity to other proteins of known function; however, as recently noted (Bork and Koonin 1998; Skolnick and Fetrow 2000), the relationship between functional similarity and sequence similarity is not straightforward. Similarly, although structural information significantly adds to our biological understanding of the protein, the relationship between structure and function is not all that clear either (Martin et al. 1998; Hegyi and Gerstein 1999).
We propose to take the functional analysis a step further, first by predicting the structure from the sequence, then by screening each predicted structure with specific three-dimensional motifs based on specific functional sites in proteins. Here, we showed that such an analysis of a eukaryotic genome for one commonly studied function significantly adds to our knowledge of protein function, creating hypotheses that can now be tested experimentally. This method allows one to verify predictions made by the sequence-based method, and use of specific structure and function information allows us to dig deeper into the sequence databases. Several novel predictions of disulfide oxidoreductases in the yeast genome and one for a C. elegans and a human sequence are presented here.
Detailed comparison of the FFF results to the Blocks motif method shows that the FFF analysis yields significantly fewer probable false-positives than the sequence-only motif method. Knowledge of the specific functional sites in even a rough protein model provides significant insight into biochemical mechanisms. This last advantage cannot be obtained by sequence-only methods for function prediction. These advantages have allowed us to make several novel predictions of disulfide oxidoreductases in the yeast genome and one for a homologous protein in the human genome. Thus, structure-based functional analysis provides additional information to the large-scale genomics projects and significantly enhances our understanding of the biological functioning of proteins expressed in each organism.
Materials and methods
Threading
The sequences from the S. cerevisiae genome were downloaded from the Saccharymyces genome database (SGD; http://genome-www.stanford.edu/ Saccharomyces). Each sequence was threaded through structures in a database of 1501 nonredundant proteins taken from the Brookhaven Protein Database. The threading algorithm used was developed by Godzik and coworkers (Jaroszewski et al. 1998). This threading algorithm analyzes the sequence-to-structure alignments by using three scoring functions, sq, br, and tt, as described (Jaroszewski et al. 1998). The six top sequences for each scoring function were analyzed for the presence of the functional site by using the FFF, as previously described (Fetrow et al. 1998).
Conservation profile
A conservation profile was calculated (Zhang et al. 1998). Each sequence was used to search the National Center for Biotechnology Information nonredundant protein sequence database (www.ncbi.nlm.nih.gov) by using Psi-BLAST (Altschul et al. 1997). All sequences with an e-score <0.01 were subject to a multiple sequence alignment and the conservation of the functional residues identified by the FFF calculated (Zhang et al. 1998). If all three functional residues were found in >50% of the aligned sequences, then the sequence is validated by the conservation profile analysis.
Sequence motif databases
In Table 1, each sequence identified by the FFF as being a disulfide oxidoreductase was submitted to the Web-based version of Prints (Attwood et al. 1997; www.biochem.ucl.ac.uk/bsm/ dbbrowser/PRINTS/PRINTS.html), Prosite (Hofmann et al. 1999; www.expasy.ch/prosite/), and Blocks (Henikoff et al. 1999; www.blocks.fhcrc.org/blocks_search.html). Default parameters were used. Sequences identified as disulfide oxidoreductases in one of the 10 top scores by these methods are marked in Table 1. For the Blocks data generated in Figure 4 ▶, the downloadable version of Blocks (BLIMPS) was used. The three Blocks motifs for the glutaredoxins and thioredoxins screened all sequences in the yeast genome.
Protein preparation
The gene corresponding to the sequence of the predicted N33 protein soluble domain was cloned by PCR from a human prostatic adenocarcinoma cDNA library (Clontech). Site-specific mutations were made to replace some or all of the non-active site cysteines with alanine, by using standard PCR methods. Various constructs were subcloned into a T7 expression vector (Novagen), and protein was overexpressed using the host BL21(DE3). The protein, expressed in inclusion bodies, was solubilized by the addition of guanidine HCl and purified by RPHPLC C4 (MeCN/TFA). Attempts made to refold the protein by using dialysis in the presence of redox buffers yielded material that either was unstructured by CD or NMR or consisted of high molecular weight aggregates. CD spectroscopy was performed on an Aviv 62DS spectrometer at 25°C, at a concentration of ∼15 μM. NMR spectra were acquired at 500 MHz on a Bruker AMXspectrometer at 25°C. Assays for activity included the DTNB assay for thioredoxin-like oxidation and the insulin assay for thioredoxin-like reduction (Karaoglu et al. 1997).
Acknowledgments
This research was supported in part by NIH Grant GM48835 of the Division of General Medical Sciences.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.49201.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Shang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood, T.K., Avison, H., Beck, M.E., Bewley, M., Bleasby, A.J., Brewster, F., Cooper, P., Degtyarenko, K., Geddes, A.J., Flower, D.R., et al. 1997. The PRINTS database of protein fingerprints: A novel information resource for computational molecular biology. J. Chem. Inf. Comput. Sci. 37 417–424. [DOI] [PubMed] [Google Scholar]
- Bork, P. and Koonin, E.V. 1998. Predicting functions from protein sequences—Where are the bottlenecks? Nat. Genet. 18 313–318. [DOI] [PubMed] [Google Scholar]
- Bushweller, J.H., Aslund, F., Wuthrich, K., and Holmgren, A. 1992. Structural and functional characterization of the mutant Escherichia coli glutaredoxin (C14-S) and its mixed disulfide with glutathione. Biochemistry 31 9288–9293. [DOI] [PubMed] [Google Scholar]
- Chaohong, S., Holmgren, A., and Bushweller, J.H. 1997. Complete 1H, 13C, and 15N NMR resonance assignments and secondary structure of human glutaredoxin in the fully reduced form. Protein Sci. 6 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson, H.J., Jeng, M.F., Tennant, L.L., Slaby, I., Lindell, M., Dui, D.S., Kuprin, S., and Holmgren, A. 1997. Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: Structural and functional characterization of mutants of Asp 26 and Lys 57. Biochemistry 36 2622–2636. [DOI] [PubMed] [Google Scholar]
- Farquhar, R., Honey, N., Murant, S.J., Bossier, P., Schultz, L., Montgomery, D., Ellis, R.W., Freedman, R.B., and Tuite, M.F. 1991. Protein disulfide isomerase is essential for viability in Saccharomyces cerevisiae. Gene 108 81–89. [DOI] [PubMed] [Google Scholar]
- Fetrow, J.S. and Skolnick, J. 1998. Method for prediction of protein function from sequence using the sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J. Mol. Biol. 281 949–968. [DOI] [PubMed] [Google Scholar]
- Fetrow, J.S., Godzik, A., and Skolnick, J. 1998. Functional analysis of the Escherichia coli genome using the sequence-to-structure-to-function paradigm: Identification of proteins exhibiting the glutaredoxin/thioredoxin disulfide oxidoreductase activity. J. Mol. Biol. 282 703–711. [DOI] [PubMed] [Google Scholar]
- Fetrow, J.S., Siew, N., and Skolnick, J. 1999. Structure-based functional motif identifies a potential disulfide oxidoreductase active site in the serine/threonine protein phosphatase-1 subfamily. FASEB J. 13 1866–1874. [DOI] [PubMed] [Google Scholar]
- Hegyi, H. and Gerstein, M. 1999. The relationship between protein structure and function: A comprehensive survey with application to the yeast genome. J. Mol. Biol. 288 147–164. [DOI] [PubMed] [Google Scholar]
- Henikoff, J.G., Henikoff, S., and Pietrokovski, S. 1999. New features of the Blocks database servers. Nucleic Acids Res. 27 226–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, K., Bucher, P., Falquet, L., and Bairoch, A. 1999. The Prosite database, its status in 1999. Nucleic Acids Res. 27 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren, A. and Bjornstedt, M. 1995. Thioredoxin and thioredoxin reductase. Methods Enzymol. 252 199–208. [DOI] [PubMed] [Google Scholar]
- Holst, B., Tachibana, C., and Winther, J.R. 1997. Active site mutations in yeast protein disulfide isomerase cause dithiothreitol sensitivity and a reduced rate of protein folding in the endoplasmic reticulum. J. Cell Biol. 138 1229–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroszewski, L., Rychlewski, L., Zhang, B., and Godzik, A. 1998. Fold prediction by a hierarchy of sequence, threading, and modeling methods. Protein Sci. 7 1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaoglu, D., Kelleher, D.J., and Gilmore, R. 1995. Functional characterization of Ost3p. Loss of the 34-kD subunit of the Saccharomyces cerevisiae oligosaccharyltransferase results in biased underglycosylation of acceptor substrates. J. Cell Biol. 130 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaoglu, D., Kelleher, D.J., and Gilmore, R 1997. The highly conserved stt3 protein is a subunit of the yeast oligosaccharyltransferase and forms a subcomplex with ost3p and ost4p. J. Biol. Chem. 272 32513–32520. [DOI] [PubMed] [Google Scholar]
- Knauer, R. and Lehle, L. 1999a. The oligosaccharyltransferase complex from Saccharomyces cerevisiae. Isolation of the ost6 gene, its synthetic interaction with ost3, and analysis of the native complex. J. Biol. Chem. 274 17249–17256. [DOI] [PubMed] [Google Scholar]
- Knauer, R. and Lehle, L. 1999b. The oligosaccharyltransferase complex in yeast. Biochim. Biophys. Acta 1426 259–273. [DOI] [PubMed] [Google Scholar]
- Kortemme, T. and Creighton, T.E. 1995. Ionisation of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol. 253 799–812. [DOI] [PubMed] [Google Scholar]
- Lathrop, R. and Smith, T.F. 1996. Global optimum protein threading with gapped alignment and empirical pair scoring function. J. Mol. Biol. 255 641–665. [DOI] [PubMed] [Google Scholar]
- Levy, A., Dang, U.C., and Bookstein, R. 1999. High-density screen of human tumor cell lines for homozygous deletions of loci on chromosome arm 8p. Genes Chromosomes Cancer 24 42–47. [DOI] [PubMed] [Google Scholar]
- MacGrogan, D., Levy, A., Bova, G.S., Isaacs, W.B., and Bookstein, R. 1996. Structure and methylation-associated silencing of a gene within a homozygously deleted region of human chromosome band 8p22. Genomics 35 55–65. [DOI] [PubMed] [Google Scholar]
- Martin, A.C., Orengo, C.A., Hutchinson, E.G., Jones, S., Karmirantzou, M., Laskowski, R.A., Mitchell, J.B., Taroni, C., and Thornton, J.M. 1998. Protein folds and functions. Structure 6 875–884. [DOI] [PubMed] [Google Scholar]
- Muller, E.G. 1992. Thioredoxin genes in Saccharomyces cerevisiae: Map positions of TRX1 and TRX2. Yeast 8 117–120. [DOI] [PubMed] [Google Scholar]
- Pedrajas, J.R., Kosmidou, E., Miranda-Vizuete, A., Gustafsson, J.A., Wright, A.P., and Spyrou, G. 1999. Identification and functional characterization of a novel mitochondrial thioredoxin system in Saccharomyces cerevisiae. J. Biol. Chem. 274 6366–6373. [DOI] [PubMed] [Google Scholar]
- Pietrovski, S., Henikoff, J.G., and Henikoff, S. 1996. The Blocks database—As system for protein classification. Nucleic Acids Res. 24 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick, J. and Fetrow, J.S. 2000. From genes to protein structure and function: Novel applications of computational approaches in the genomic era. Trends Biotechnol. 18 34–39. [DOI] [PubMed] [Google Scholar]
- Skolnick, J., Fetrow, J.S., and Kolinski, A. 2000. Structural genomics and its importance for gene function analysis. Nat. Biotechnol. 18 283–287. [DOI] [PubMed] [Google Scholar]
- Tachibana, C. and Stevens, T.H. 1992. The yeast EUG1 gene encodes an endoplasmic reticulum protein that is functionally related to protein disulfide ixomerase. Mol. Cell. Biol. 12 4601–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa, H., Miura, T., Katakura, Y., and Mizunaga, T. 1991. Molecular structure of a yeast gene, PDI1, encoding protein disulfide isomerase that is essential for cell growth. J. Biochem. (Tokyo) 110 306–313. [DOI] [PubMed] [Google Scholar]
- Tachikawa, H., Funahashi, W., Takeuchi, Y., Nakanisi, H., Nishihara, R., Katoh, S., Gao, X.D., Mizunaga, T., and Fujimoto, D. 1997. Overproduction of Mpd2p suppresses the lethality of protein disulfide isomerase depletion in a CXXC sequence dependent manner. Biochem. Biophys. Res. Comm. 239 710–714. [DOI] [PubMed] [Google Scholar]
- Walker, K.W., Lyles, M.M., and Gilbert, H.F. 1996. Catalysis of oxidative protein folding by mutants of protein disulfide isomerase with a single active-site cysteine. Biochemistry 35 1972–1980. [DOI] [PubMed] [Google Scholar]
- Wallace, A.C., Birkakoti, N., and Thornton, J.M. 1997. TESS: A geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases: Application to enzyme active sites. Protein Sci. 6 2308–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal, V., Darby, N.J., and Winther, J.R. 1999. Functional properties of the two redox-active sites in yeast protein disulphide isomerase in vitro and in vivo. J. Mol. Biol. 286 1229–1239. [DOI] [PubMed] [Google Scholar]
- Yang, Y.F. and Wells, W.W. 1991a. Catalytic mechanism of thioltransferase. J. Biol. Chem. 266 12766–12771. [PubMed] [Google Scholar]
- Yang, Y.F. and Wells, W.W. 1991b. Identification and characterization of the functional amino acids at the active center of pig liver thioltransferase by site-directed mutagenesis. J. Biol. Chem. 266 12759–12765. [PubMed] [Google Scholar]
- Yang, Y., Jao, S., Nanduri, S., Starke, D.W., Mieyal, J.J., and Qin, J. 1998. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, and C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry 37 17145–17156. [DOI] [PubMed] [Google Scholar]
- Zhang, L., Godzik, A., Skolnick, J., and Fetrow, J.S. 1998. Functional analysis of E. coli proteins for members of the a/b hydrolase family. Fold. Des. 3 535–548. [DOI] [PubMed] [Google Scholar]