

Abstract
The SH3 domain of Eps8 was previously found to form an intertwined, domain-swapped dimer. We report here a monomeric structure of the EPS8 SH3 domain obtained from crystals grown at low pH, as well as an improved domain-swapped dimer structure at 1.8 Å resolution. In the domain-swapped dimer the asymmetric unit contains two "hybrid-monomers." In the low pH form there are two independently folded SH3 molecules per asymmetric unit. The formation of intermolecular salt bridges is thought to be the reason for the formation of the dimer. On the basis of the monomer SH3 structure, it is argued that Eps8 SH3 should, in principle, bind to peptides containing a PxxP motif. Recently it was reported that Eps8 SH3 binds to a peptide with a PxxDY motif. Because the "SH3 fold" is conserved, alternate binding sites may be possible for the PxxDY motif to bind. The strand exchange or domain swap occurs at the n-src loops because the n-src loops are flexible. The thermal b-factors also indicate the flexible nature of n-src loops and a possible handle for domain swap initiation. Despite the loop swapping, the typical SH3 fold in both forms is conserved structurally. The interface of the acidic form of SH3 is stabilized by a tetragonal network of water molecules above hydrophobic residues. The intertwined dimer interface is stabilized by hydrophobic and aromatic stacking interactions in the core and by hydrophilic interactions on the surface.
Keywords: SH3 domains, Eps8, domain swapping, intertwined dimer, salt bridge
Many signaling molecules are modular in nature and some of the modules facilitate protein–protein interactions (Pawson 1995). Src homology 2 and 3 (SH2 and SH3) domains are examples of modular domains that participate in a variety of protein–protein interactions (Bork et al. 1997). SH2 domains recognize molecules containing phosphotyrosine residues and SH3 domains recognize proteins with polyproline stretches, in particular a PxxP sequence motif with a positively charged residue located one or two amino acids away from the PxxP motif (Wittekind et al. 1994). In addition to intermolecular interactions, SH3 domains may provide a mechanism for intramolecular interactions: SH3 domains have been shown to bind intramolecularly to sequences that contain a single proline residue (Sicheri et al. 1997; Xu et al. 1997). In fact, the PxxP motif is responsible for maintaining the polyproline II (PPII) helix, in which hydrophobic proline residues line up in the interdigitated binding grooves (Lim and Richards 1994). Once the basic PPII helix is provided, SH3 domains bind even to peptides with nonnatural N-substituted residues with higher affinities (Nguyen et al. 1998).
The binding of polyproline peptides to SH3 domains has been observed in two possible orientations, class I and class II (Feng et al. 1994). A positively charged residue, flanking the PxxP motif at either the N or C terminus, is the anchoring residue that determines the direction of peptide binding (Feng et al. 1994; Lim et al. 1994; Wu et al. 1995). The three-dimensional structures of many SH3 domains have been determined to atomic resolution either by nuclear magnetic resonance (NMR) or by X-ray crystallographic techniques (Kishan et al. 1997). More structures have been determined in the last 3 yr: human nebulin (Politou et al. 1998), rat amphiphysin 2 (Owen et al. 1998), human hck (Sicheri et al. 1997), and an SH3 domain from bruton's tyrosine kinase (Hansson et al. 1998). A typical SH3 domain (Fig. 1 ▶) is formed by two orthogonal β-sheets, each sheet formed by three antiparallel β-strands. The loops connecting the β-strands (B1–B5) are sequentially named as the RT, n-src, and distal loops. The last loop connecting strand B4 with strand B5 is instead a short 310-helix, generally comprising three amino acids in most SH3 structures. Of all the structures, the SH3 domain structure from epidermal growth factor receptor pathway substrate 8 (Eps8) is novel, because it forms a strand-exchanged intertwined dimer (Kishan et al. 1997). The n-src loop of the Eps8 SH3 polypeptide chain has a unique conformation compared with other SH3 domains. Instead of folding back onto its own polypeptide chain to form an SH3 molecule, it extends into a neighboring molecule to make half of the other SH3 molecule. The "swapping" between two partners of a dimer is cooperative, and, as a result, two polypeptide chains of SH3 molecules fold into an intertwined dimer (Fig. 1a ▶ of Kishan et al. 1997; Fig. 2B ▶). The main consequence of Eps8 SH3 intertwined dimer formation is that it obscures the polyproline peptide binding groove of both partners of the SH3 dimer. The peptide binding groove is formed between the RT-loop and n-src loop and consists of hydrophobic residues (Fig. 1 ▶). All of the residues that form the peptide binding groove are more or less conserved in most of the SH3 domains. In this paper we report the crystal structures of an improved Eps8 SH3 dimer model (hereafter called DSH3) refined to 1.8 Å, and an Eps8 SH3 monomer (hereafter called MSH3) at 2.0 Å resolution. A 2.5 Å structure of DSH3 has been previously described (Kishan et al. 1997).
Fig. 1.

Schematic diagram of SH3 fold. The β-strands are labeled as B1 to B5 and are color coded (blue and green), indicating the strands separated because of swapping. The hinge loop is drawn in red. The N terminus, C terminus, RT-loop, n-src loop, and distal loop are marked; the 310-helix is labeled as H1. The conserved residues responsible for polyproline binding are shown and labeled.
Fig. 2.
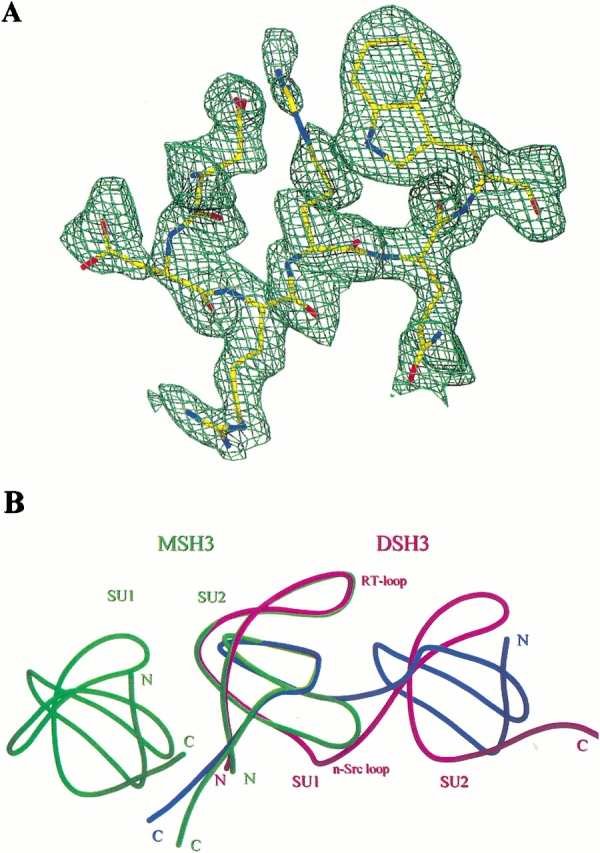
(A) Omit electron density for n-src loop (residues 35–40) of chain A (DSH3), calculated without the loop during refinement (see Materials and Methods). The map was contoured at a 1.0 level of sigma. This figure was generated with Molray (http://portray.bmc.uu.se/cgi-bin/markh/molray.pl) and rendered with Povray (http://www.povray.org/). (B) Quaternary structure of DSH3 (magenta, blue) and MSH3 (green). The relative positions of folded SH3 molecules were compared. Subunits 1 and 2 (SU1 and SU2) for both DSH3 and MSH3 are labeled. The N terminus, C terminus, RT-loop, and n-src loops are shown. Note the strand exchange at the n-src loops.
Results
Molecular structure and packing
The crystals of DSH3 and MSH3 were grown at pH 7.0 and 4.0, respectively, from a test set of 96 solutions of Crystal screen (Hampton Research Corp.). DSH3 crystallized in the P1 space group and MSH3 in P31. In both cases two SH3 molecules are present per asymmetric unit. DSH3 was refined to 1.8 Å and MSH3 to 2.0 Å with good R-factors and R-free factors. The root mean square (RMS) bond length and angle deviations during refinement were within the allowed range for both structures (Table 1). The Ramachandran plot of both molecules shows that all of the residues have φ/Ψ angles within allowed regions (data not shown). In DSH3 the two polypeptides exchange their strands, forming an intertwined dimer. Each SH3 structural motif is formed by two polypeptide chains (amino acids 1–35 from chain A and 40–64 from chain B and vice versa). The n-src loop residues 36–39 swap the polypeptide chains to complete the folding. The SH3 domain formed in this way could be termed a "hybrid-monomer" because it is formed by two polypeptide chains. MSH3 has two SH3 molecules in the asymmetric unit and each polypeptide folds into an independent SH3 molecule. The Matthews' coefficients (Matthews 1985) are approximately similar for both DSH3 and MSH3 (1.90 Å3/D and 1.85 Å3/D, respectively). The solvent contents for DSH3 and MSH3 in the unit cell are approximately the same (∼52%), and in both instances the packing of molecules in the unit cell is exceptionally tight when compared with other crystal packings. Formation of an intertwined dimer in DSH3 results in ∼1700 Å2 being the total buried surface area. The two molecules of MSH3, compared with DSH3, pack with a significantly smaller interface: 1100 Å2 is buried in the noncrystal symmetric dimer. The DSH3 dimer has two polypeptides intertwined. Although each polypeptide chain would not form a completely folded protein in its current conformation, by intertwined dimerization each polypeptide chain buries more surface area. When the interface between the two individual polypeptide chains A and B of DSH3 are calculated, ∼4000 Å2 surface area is buried.
Table 1.
Data collection and refinement statistics
| Space group | P1 | P31 |
| Cell dimensions (Å) (°) | a = 27.9 | a = 49.7 |
| b = 28.5 | b = 49.7 | |
| c = 37.2 | c = 36.5 | |
| α = 107.4 | α = 90.0 | |
| β = 96.9 | β = 90.0 | |
| γ = 104.6 | γ = 120.0 | |
| Molecules/asymmetric unit | 2 | 2 |
| Resolution (Å) | 1.8 | 2.0 |
| Completeness (%) | 94.0 | 99.7 |
| Observed reflections | 33871 | 24299 |
| Unique reflections | 9025 | 6795 |
| Rmergea (%) | 7.2 | 6.9 |
| R-factorb (%) | 20.2 | 18.9 |
| R-free (%) | 23.3 | 23.5 |
| Number of protein atoms | 970 | 963 |
| Number of water molecules | 94 | 92 |
| Average B-value (Å2) | ||
| overall | 20.8 | 24.6 |
| main chain | 19.2 | 23.9 |
| side chain | 22.3 | 25.4 |
| R.m.s.d. bond lengths (Å) | 0.007 | 0.016 |
| R.m.s.d. bond angles (°) | 1.38 | 1.85 |
a Rmerge = ∑|Ii − 〈I〉| Σ 〈I〉.
bR-factor = ∑|Fo = Fc|/∑Fo.
Structure comparisons
The higher-resolution SH3 dimer structure (DSH3) is essentially the same as the 2.5 Å resolution structure (Kishan et al. 1997, PDB code 1AOJ) with a 0.57 Å overall RMS deviation for main chain atoms. The only significant deviations are at the n-src loop, which differ by up to 2.6 Å. This is presumably a result of the poor electron density for the n-src loops in the lower resolution structure. Although in both the structures the n-src loops have high thermal b-factors, the electron density for the loops in the DSH3 structure is better defined in the 1.8 Å structure (Fig. 2A ▶). The two polypeptide chains of DSH3 are equally similar in structure with an overall RMS deviation for the main chain atoms of 0.6 Å. Major deviations are observed at RT- and distal loops. The maximum deviation of about 1.7 Å at the RT-loop is due to two asymmetric hydrogen bonds between Ser 21A (for residue Ser 21 of chain A) OG and Ser 21B main chain oxygen (OG to O distance 3.1 Å) and Ser 20A main chain oxygen and Ser 21B OG through a water molecule (267 OH2). The differences of ∼1.0 Å between the distal loops may be due to the differences in the crystallographic environment between the two loops. The two molecules in MSH3 asymmetric unit are arranged as a dimer with a quaternary structure much different from that observed for DSH3, even if one were to model the DSH3 dimer without the strand exchange (Fig. 2B ▶). The two MSH3 molecules are similar to each other, with a main chain RMS deviation of 0.14 Å. Therefore, for all comparisons only one of the molecules (Chain A) is chosen.
The comparison of either of the hybrid monomers of DSH3 with either of the monomers of MSH3 shows a very high structural alignment except at the n-src loop and distalloop (Fig. 2B ▶). For comparison, the n-src loop region of the hybrid-monomer (DSH3) and the corresponding residues in the MSH3 were not taken into consideration. Because the secondary structure and overall "SH3 fold" is highly conserved and the n-src loop is responsible for chain swapping, the φ/Ψ angles of the residues in n-src loops are compared between DSH3 and MSH3. After visual inspection of both the aligned loops, it is apparent that three main-chain dihedral angles are important in bringing about the chain swapping. These are Leu 34 Ψ (a difference of ∼210° between DSH3 and MSH3), Arg 37 φ (∼140°) and Gln 39 Ψ (∼180°). After applying these differences to MSH3, the chain progression is directionally similar to that of DSH3, although not identical. Mongiovi et al. (1999) have shown that Eps8 SH3 exists in dynamic equilibrium and the dimer and monomer species can be separated on a fast protein liquid chromatography system. However, when peaks corresponding to the monomers and dimers were collected and individually subjected to gel-filtration chromatography, they gave both dimeric as well as monomeric peaks. If this equilibrium has to be maintained, in addition to the changes in φ/Ψ angles described, a considerable number of noncovalent bonds must be broken and formed. Main-chain:main-chain hydrogen bonding is much more extensive in MSH3 than in DSH3 (data not shown).
The average temperature factors of main chain atoms in both DSH3 and MSH3 are moderate (about 19 Å2 and 24 Å2, respectively). DSH3 has higher average thermal b-factors for main chain atoms in the n-src and distal-loops. MSH3 has higher b-factors from its N terminal to the end of strand B2 (which includes the RT-loop), than does the rest of the molecule, which has much lower-than-average main chain b-factors (Fig. 3 ▶).
Fig. 3.

Thermal b-factor comparison of DSH3 and MSH3. The strands B1 to B5 and the 310-helix are shown as horizontal bars appropriate to their position. The region where the strand exchange (n-src loop) takes place is indicated with an arrow.
Salt bridges
A number of salt bridges formed in both DSH3 and in MSH3. There are no salt bridges between symmetry-related molecules in DSH3, whereas there are two such bridges in MSH3. It is interesting to note that four Glu residues in the intertwined dimer participate in salt-bridge formation. In MSH3, only one Glu residue is involved in salt bridging. Both Glu 22 residues from chain A and B are involved in a network of salt bridges that connect the two hybrid monomers of the intertwined dimer. The environment in which both Glu 22 residues of DSH3 form salt bridges is more or less similar. In chain A, the NH1 and NH2 atoms of Arg 18 are juxtaposed against OE1 and OE2 of Glu 22. In addition, Glu 22 forms a salt link with Lys 42 in both chains (distances between OE2 of Glu 22 and NZ of Lys 42 in chains A and B are 2.8 and 2.5 Å, respectively; Fig. 4A ▶). In chain B, the NE and NH1 atoms of Arg 18 make interactions with the Glu 22 side chain. The other side of the Arg 18 guanidyl group makes another salt bridge with Asp 35 (Fig. 4A ▶). However, the environment of the Glu 22 residues in chains A and B is different in MSH3. Both Glu 22 residues in MSH3 are involved in forming hydrogen bonds with Asp 49 from symmetry-related molecules of the same corresponding chains. Glu 32 in MSH3 (chains A and B) forms a weak interaction with Arg 44. This interaction is not expected because at pH 4.0 Glu 32 is supposed to be protonated. Glu 32 of both chains in DSH3 forms charge–charge interactions with N-terminal Lys 6. Two other glutamates, Glu 65 in chains A and B, were having very poor electron density in DSH3 as well as in MSH3. Therefore they were not included in the model. Only 2 of 14 Asp residues from both chains are involved in salt bridging in DSH3. It is interesting to note that in MSH3 the major participation in salt-bridge formation is from 9 of 14 Asp residues. The Asp 35 is the only common residue involved in ionic interactions within the n-src loops of both DSH3 and MSH3. However, the other participating residue in the salt bridge is different. In DSH3 Asp 35 residues interact with Arg 38 residues. However, in MSH3 Asp 35 residues make salt bridges with Arg 37 residues. The change in these ionic interactions could lead to differences in the conformations of n-src loops because both Asp 35 and Arg 37 are in the n-src loops (Fig 4B ▶).
Fig. 4.
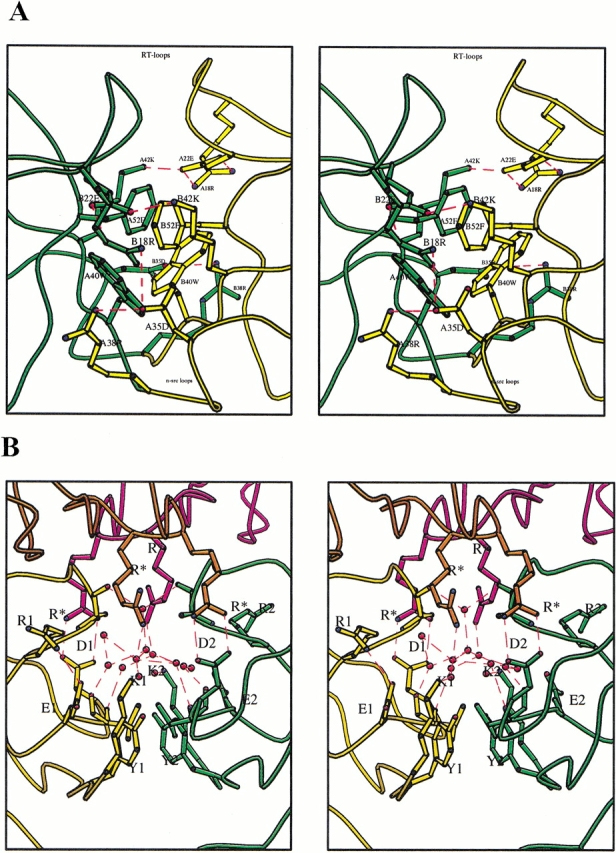
(A) Hydrophobic interface of hybrid-monomers in DSH3. The two molecules are shown with different colors (gold and green). The aromatic stacking and edge-to-face interactions of aromatic rings can be visualized. The salt bridge stabilization of the interface is shown. Note the salt links formed by A22E and B22E with A42K and B42K stabilizing at the RT-loop region. Salt bridges are drawn as dashed lines in red. The positions of the RT-loop and n-src loops are indicated. Residues are labeled with chain identifier, residue number, and single-letter code for amino acids (e.g., A35D for Asp 35 from chain A). (B) Water channel at the interface of the two molecules in MSH3. The two chains are color coded (green and gold). Symmetry-related molecules are color coded as orange and magenta. Waters are shown as red circles and water bridges are shown with their contacting polar amino acids as red dashed lines. The salt bridges are shown as red dashed lines. (R*) Arg from symmetry-related molecules; (K1) Lys 6; (Y1) Tyr 8; (D1) Asp 29; (E1) Glu 32; (R1) Arg 44 from chain A. The corresponding single-letter codes with numeric 2 are for chain B.
Water structure
There are approximately the same number of water molecules found in DSH3 and MSH3 asymmetric units, and two-thirds of water molecules have conserved positions, that is, either they occupy approximately the same location in both structures or are hydrogen bonded to the same amino acid. The average thermal b-factors for waters in both the crystal structures are nearly the same (∼36 Å2). The polyproline binding groove is covered by a network of waters in all of the molecules of DSH3 and MSH3 at a distance slightly above the van der Waals' distance between water molecules and the hydrophobic atoms. The network is somewhat conserved in both the molecules of DSH3 and one of the two molecules of MSH3 (chain B). This network of water molecules could be called "water bridges" because two or more water molecules are involved in connecting two residues over a distance of a few angstroms. In DSH3 the water bridge that is formed binds the two chains overshadowing the polyproline binding groove. The presence of water molecules at van der Waals' distance to apolar side chains is not a new phenomenon. Thanki et al. (1988) showed that water molecules could be observed at van der Waals' distance to apolar side chains, provided they make at least one contact with polar atoms. Besides the water bridges, there is an additional interesting water network observed in MSH3. The two monomers in the MSH3 asymmetric unit have an interface that is partly hydrophobic and partly hydrophilic. At the interface, 13 waters form a water network (Fig. 4B ▶). Of the 13 water molecules, 10 make hydrogen bonds with polar atoms of the protein, and 3 waters are bound to other waters in the network. In such a situation, hydrogen bonds are formed between four atoms in the form of tetragonal shapes. The water network covers the hydrophobic interface of the two monomers. The shape of the network is visualized as tetragonal rings. Such water bridges are not observed frequently in protein structures; however, they have been seen in the hydrophobic intermolecular interface of crambin (Teeter 1984). Two symmetry-related molecules interact with both the molecules of MSH3 above this water network, causing it to appear like a cavity filled with water molecules.
Discussion
Many proteins are known to form strand intertwined dimers (Schlunegger et al. 1997). Eps8 SH3 follows the proteins found earlier to be of this kind. It is crystallized in two crystal forms: in one of the forms it is a strand exchanged dimer and in the other it is a "closed monomer" as defined by Schlunegger et al. (1997). The structure and fold is completely conserved when only the monomeric part of the molecules is considered. The only deviations in the intertwined dimer are at the n-src loop where the strand swap takes place (Fig. 2B ▶). As reported earlier (Kishan et al. 1997) and also thorough investigations by Mongiovi et al. (1999), in solution both monomer and swapped dimers are in dynamic equilibrium, with the monomer being the major species. In crystals these two forms are trapped, one at pH 7.0 (DSH3) and another at pH 4.0 (MSH3). However, to maintain the equilibrium between the monomer and the dimer, the molecule has to cross a large energy barrier (Schlunegger et al. 1997). For the conversion of DSH3 into MSH3, three torsion angles of the n-src loop have to be rotated extensively. While the n-src loop changes its conformation to become MSH3 from DSH3 or vice versa, the polypeptide on either side of the n-src loop has to reorient considerably. In this process, various interactions in the C interface (as described in Schlunegger et al. 1997), namely, hydrophobic, ionic, and polar interactions, have to be disrupted. It was suggested by Schlunegger et al. (1997) that the length of the hinge region (i.e., the n-src loop) might be another factor that could influence the swap. The n-src loop in Eps8 SH3 is one of the shortest n-src loops among many known SH3 structures.
The pH difference between the two crystal forms and exposure of hydrophobic surfaces are a likely explanation for the fact that we are able to observe both the monomeric and dimeric forms of Eps8 SH3. In DSH3 the core of the interface between the two hybrid monomers is hydrophobic, and it is surrounded by polar interactions as well as by salt bridges involving Glu residues on the surface (Fig 4A ▶). It was shown by Murray et al. (1998) that exposed hydrophobic groups could be a reason for domain swapping in CD2. The hydrophobic surface formed by Trp 40 and Phe 52 is well stabilized by the intertwined dimer formation through aromatic stacking interactions from the same region of the partner molecule (Fig. 4A ▶). Although the same hydrophobic surface in MSH3 is involved in crystal contacts, the interface is further stabilized by polar interactions surrounding the aromatic residues (data not shown). The pK values of Asp and Glu side chains are 3.5–4.0 and 4.3, respectively. When the pH was lowered to 4.0, depending on the microenvironment around the Glu residues, they either become protonated or are still charged. Because Glu 22 residues from both chain A and B of DSH3 (Fig. 4A ▶) are responsible for the intermolecular salt bridges, by lowering the pH it is likely that the salt bridges will be disrupted and that monomer formation will be favored. We do not yet know how a single Glu residue could change the dynamics of intertwined dimer and monomer equilibrium. Therefore, when triggered by the disruption of a stable salt link supported by favorable hydrophobic and polar interactions, DSH3 could attain the conformation of MSH3.
The difference in the microenvironment around Glu 22 and Glu 32 is evidenced by formation of a weak salt link between Glu32A and Arg44A with a distance of 3.1 Å (the corresponding distance in the B chain is 3.8 Å). This is the only interaction in MSH3 where a Glu participates in a salt bridge. A close look at the vicinity of all of the Asp and Glu residues in MSH3 shows that, although there is a possibility for salt bridge formation, side chains of Lys and Arg residues prefer to be away from Glu. For example, Arg 18A makes a salt bridge with Glu 22A in DSH3. However, the same salt bridge is replaced by a salt link interaction between Arg 18A and Asp 14B of a symmetry-related molecule in MSH3. The main chain conformation at Arg 18A is the same for both DSH3 and MSH3. Only the side chain conformation is changed to facilitate the new salt bridge between the Arg and Asp. The electron density for these residues is well-defined and the thermal b-factors are also low to moderate. Both Glu 22A and Glu 22B of MSH3 form hydrogen bonds (∼2.8 Å) with Asp 49A and Asp 49B, respectively. This kind of hydrogen bond is possible only with protonated or neutral Glu residues.
The molecular environment of the dimer interfaces for DSH3 and MSH3 is not the same. The relative orientation of the two molecules of both dimers is different, even when symmetry-related molecules are considered. Differences in molecular packing in different space groups for the same protein is not uncommon. For example, the enzyme trypanosomal triosephosphate isomerase was crystallized into three different crystal forms and has four packing environments (Kishan et al. 1994). The interface of the two molecules in DSH3 is more or less as described previously (Kishan et al. 1997). It has to be noted that as a result of strand swapping, hydrophobic interactions, and charge–charge interactions, the dimer interface in DSH3 resembles the central core of a globular protein (Fig. 4A ▶). The two molecules of MSH3 (in the asymmetric dimer) are much less tightly packed and the interface of the molecules buries few hydrophobic residues. There are no strong charge–charge interactions to support the core. Instead, the interface is like a water channel with 13 waters forming a water network above the hydrophobic interface interacting with the polar side chains. This water network is stabilized by two symmetry-related molecules above this water channel, which makes it a cavity filled with water molecules (Fig 4B ▶). The overall folding and stability of a protein depends on various interactions. DSH3 is stabilized by all of the interactions at the interface that could be expected in a folded protein.
The thermal b-factors could be taken as indicators of the flexibility of n-src loop. The presence of proper electron density for all of the main chain atoms and most of the side chain atoms in both DSH3 as well as MSH3 is an indication of no ambiguity in the propensity of the chain at the n-src loop. As mentioned earlier (Kishan et al. 1997), in many of the SH3 structures available, the n-src loop has high b-factors in the case of X-ray crystallographically derived structures or large RMS deviations for multiple structures derived by the NMR method. This indicates a tendency for the n-src loop to be flexible and suggests multiple conformations. Flexible loops may start swapping of the peptide chains, provided the other conditions are favorable. In Eps8 SH3 the flexibility of the n-src loops could be one of the reasons for the formation of an intertwined dimer. It is possible that, once Glu 22 ionic interactions with Lys 42 and Arg 18 are disrupted, both the RT-loops cannot hold the same conformation as that depicted in Fig. 4A ▶. Consequently, the hydrophobic interface would be exposed to solvent. When the dimer becomes unstable, it will look for alternate conformations. In such a situation the n-src loop could serve as a handle to bring about the dimer–monomer interconversion. Surprisingly, MSH3 has very low b-factors for the n-src loop (Fig. 3 ▶). The observation that b-factors are rather high for the strands B1, B2, and RT-loop and distinctly lower for the rest of the molecule in both monomers of MSH3 may indicate a flexible nature for half of the molecule, facilitating a swap with another monomer to form a dimer.
From the comparison of MSH3, as well as the hybrid-monomer of DSH3, with other SH3 structures, the overall RMS differences in the C-alpha positions show that the overall structure is conserved and even the side-chain orientations are also generally conserved. In principle then, Eps8 SH3 should be able to bind to the PxxP motif. Mongiovi et al. (1999) have shown that the consensus peptide sequence that binds the Eps8 SH3 has a PxxDY motif and not the usual PxxP motif. The overall differences in the structure of Eps8 SH3 with that of any other SH3 domain are minimal (1.04 Å). One of the major differences in the sequence is the mutation of Phe/Tyr to Ile at position 57 (according to Eps8 SH3 domain numbering) (Fig. 5 ▶). Mongiovi et al. (1999) ruled out the possibility of this residue being responsible for dimerization and also showed that it is an important residue responsible for the binding of the PxxDY motif. In fact they showed that changing the Ile to Tyr at position 57 increased the binding of the PxxDY motif to Eps8 SH3 by more than 10-fold. When we translate the sequence differences into structure, there is a significant difference in the context of the consensus PxxP motif-binding region of the SH3 molecule. The Tyr residue not only defines the P0 site (Lim et al. 1994), but also binds to the main chain oxygen of the ligand peptide through its phenolic OH group. Therefore, for anchoring of a proline into the P0 site, the presence of Tyr would appear to be preferred. Although Phe (lacking only the hydroxyl) can substitute well for Tyr, an Ile at this position would affect significantly the contours of the P0 binding pocket. Therefore, one needs to examine whether any of the other differences bring about changes on the surface of Eps8 SH3 that could facilitate binding to the PxxDY motif. From the sequence alignment (Fig. 5 ▶), the differences visible are mainly in two arginines in the n-src loop, which is one of the important loops determining the peptide binding. These arginines may provide an alternate binding groove, which is different from the usual PxxP motif-binding groove. The PxxDY motif contains an Asp, a potential amino acid for salt bridging with Arg in the n-src loop. Vidal et al. (1998) suggested a second peptide binding site for N-terminal Grb2 SH3. Therefore, it will be interesting to see how Eps8 SH3 interacts with peptides containing the PxxDY motif. An Eps8 SH3 structure with the PxxDY peptide bound will throw light on this interesting behavior of Eps8 SH3.
Fig. 5.

Sequence alignment of SH3 domains whose three-dimensional structure are known either by X-ray or nuclear magnetic resonance techniques. Completely conserved W and P are shown with a dark background. The boxed columns indicate generously conserved amino acids. The secondary structure information is given at the top; β-strands are labeled B1 to B5 and the 310-helix is labeled H1. RT-loop, n-src loop (nL), and distal loops (dL) are labeled. (chspec) Chicken spectrin; (drosem5) drosophila sem-5; (hugrb2c) human growth factor receptor bound protein 2 C-terminal domain; (raapsin) rat amphiphysin; (hupi3k) human phosphotidyl inositol-3-kinase; (moueps8) mouse eps8; (hugrb2n) human growth factor receptor bound protein 2 N-terminal domain; (hucsk) human c-src kinase; (mouccrkn) mouse c-crk N-terminal domain; (hubtk) human bruton tyrosine kinase; (chsrc) chicken src; (hufyn) human fyn; (huhck) human hck; (hulck) human lck; (huneb) human nebulin; (mouabl) mouse abl; (huplcg) human plc-gamma.
Materials and methods
Mouse Eps8 SH3 expression and purification was performed as reported earlier (Kishan et al. 1997). Crystals of DSH3 were grown from hanging drops suspended over wells containing 100 mM MgCl2, 100 mM sodium formate, and 100 mM Tris (pH 7.0). Protein at 7–10 mg/mL was mixed with an equal volume of well solution. Crystals of MSH3 were grown from hanging drops suspended over wells containing 21% PEG-4000, 10% isopropanol, and 100 mM sodium citrate (pH 4.0). For the hanging drop, protein at 14 mg/mL was mixed with an equal volume of well solution.
Data were collected on a Raxis IV imageplate detector mounted on a Rigaku RU-200 rotating anode. DSH3 crystals diffracted to 1.8 Å and MSH3 crystals to 2.0 Å. They are of space group P1 and P31 respectively. The statistics of data collection and refinement are listed in Table 1. The cell dimensions for the P1 cell are 27.90 Å, 28.52 Å, 37.20 Å, 107.4°, 96.9°, and 104.6° and the Vm is 1.90 Å3/D for two monomers per asymmetric unit. Because the structure is an improvement over the previously determined structure at 2.5 Å resolution, the coordinates from PDB (http://www.rcsb.org/) code 1AOJ were used as a starting model. The minor changes in the cell were corrected and refinement was started with refinement protocols in a crystallography and NMR system (CNS, Brünger et al. 1998). A test set of 6.2% of the total reflections was used to calculate the R-free (Brünger 1992). A simulated annealing protocol was used initially to a maximum of 2000°K and later individual b-factor refinement was performed with CNS. During refinement, noncrystallographic symmetry restraints were not used. Data were used between 26–1.8 Å with a sigma cutoff of 2.0. The model was built graphically with the help of an "O" program (Jones et al. 1991). Water molecules were selected from Fo-Fc maps, which had a corresponding electron density in 2Fo-Fc maps as well, with the requirement that the waters make contact with a protein atom or another water molecule. The refinement was stopped when the R-factor and R-free were converged. The final R-factor and R-free are 20.2 and 23.4, respectively. To test whether the swapping of n-src loop has really occurred, we modeled the n-src loops of the swapped dimer to obtain two SH3 monomer molecules, as if the swapping did not occur. One cycle of simulated annealing refinement was performed on this coordinate set. This is like omit-map refinement for the n-src loop. The R-factor and R-free increased to 25.8 and 28.9, respectively, and the electron density for the n-src loop was clearly visible in the swapped conformation. The overall quality of the structure was validated by the program PROCHECK (Laskowski et al. 1993). There are no φ/Ψ angles in the disallowed region of the Ramachandran plot. The dihedral angle G-factors also are within the allowed range.
The cell dimensions of the P31 form are 49.7 Å, 49.7 Å, 36.5 Å, 90.0°, 90.0°, and 120.0°. The Vm is 1.85 Å3/D for two molecules per asymmetric unit. A test set of 10.9% of reflections was kept for R-free calculation. The data were processed and indexed with DENZO (Otwinowski and Minor 1997). The space group was initially thought to be P3121 and the Rsym for processing in this space group was not significantly different from the Rsym for P31 processing. Consequently, the original rotation function searches were performed for a monomer in P3121. A monomeric search model, constructed from a hybrid-monomer of DSH3, served as the search model. Rotation and translation functions were calculated with AmoRe, as implemented in the CCP4 package ( The CCP4 suite 1994). The rotation function revealed a clear peak at 4 sigma, and the highest peak in the corresponding translation function had a correlation coefficient of 37.2 and an R-factor of 52.1. After refinement of the position in a six-dimensional search, the correlation coefficient increased to 46.2 and the R-factor dropped to 48.8. A map calculated with the positioned model had clear interpretable electron density for the region of the n-src loop that was not included in the search model. After several cycles of model building and refinement, it became apparent the crystal space group may be of lower symmetry (P31) with a dimer in the asymmetric unit, as a perfect twofold was not possible for the positions of the side chains at the interface. A dimer model was generated. A rigid body refinement in space group P31 resulted in a shift in one monomer, and a lowering of both the R-factor and the R-free. Further refinement was performed as space group P31 with two monomers in the asymmetric unit. During refinement, data between 30–2.0 Å were used with no sigma cutoff. Simulated annealing and subsequently individual b-factor refinement were performed with CNS as described earlier. Both the molecules in the asymmetric unit were refined without noncrystallographic symmetry restraints. Waters were picked up from Fo-Fc maps, which were having consistent electron density in the 2Fo-Fc maps also. The final R-factor and R-free are 0.189 and 0.235, respectively. The structure validation with PROCHECK shows no φ/Ψ angles in the disallowed region of the Ramachandran plot. The dihedral angle G-factors are also within the allowed range.
CCP4 package and O programs (Jones et al. 1991) were used to calculate intermolecular distances, intramolecular distances, Matthews' coefficients and surface calculations. Figures were generated with Molscript (Kraulis 1991), Bobscript (Esnouf 1997), O, and ClustalX/ESPript (Thompson et al. 1994; Gouet et al. 1999). The coordinates have been deposited with Protein Data Bank at RCSB (http://www.rcsb.org/). The codes for DSH3 and MSH3 are 1I07 and 1I0C, respectively.
Acknowledgments
We thank Professor Paolo di Fiore for a gift of the Eps8 SH3 clone and one of the referees for suggesting important references.
The publication costs of this article were defrayed in part by payment of page charges.This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/
References
- Bork, P., Schultz, J., and Ponting, C.P. 1997. Cytoplasmic signalling domains: The next generation. Trends Biochem. Sci. 22 296–298. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. The free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 335 472–474. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., Delano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J-S., Kuszewski, J., Nilges, M., Pannu, et al. 1998. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. D54 905–921. [DOI] [PubMed] [Google Scholar]
- The CCP4 suite: Programs for protein crystallography Number 4 Collaborative Computional Project. 1994. Acta Crystallogr. D50 760–763. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. 15 133–138. [DOI] [PubMed] [Google Scholar]
- Feng, S., Chen, J.K., Yu, H., Simon, J.A., and Schreiber, S.L. 1994. Two binding orientations for peptides to the Src SH3 domain: Development of a general model for SH3-ligand interactions. Science 266 1241–1247. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D.I., and Metoz, F. 1999. ESPript: Multiple sequence alignments in PostScript. Bioinformatics 15 305–308. [DOI] [PubMed] [Google Scholar]
- Hansson, H., Mattsson, P.T., Allard, P., Haapaniemi, P., Vihinen, M., Smith, C.I., and Hard, T. 1998. Solution structure of the SH3 domain from Bruton's tyrosine kinase. Biochemistry 37 2912–2924. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Kishan, K.V.R., Zeelen, J.P., Noble, M.E.M., Borchert, T.V., and Wierenga, R.K. 1994. Comparison of the structures and the crystal contacts of trypanosomal triosephosphate isomerase in four different crystal forms. Protein Sci. 3 779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishan, K.V.R., Scita, G., Wong, W.T., Fiore, P.P.D., and Newcomer, M.E. 1997. The SH3 domain of eps8 exists as a novel intertwined dimer. Nat. Struct. Biol. 4 739–743. [DOI] [PubMed] [Google Scholar]
- Kraulis, P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Lim, W.A. and Richards, F.M. 1994. Critical residues in an SH3 domain from Sem-5 suggest a mechanism for proline-rich peptide recognition. Nat. Struct. Biol. 1 221–225. [DOI] [PubMed] [Google Scholar]
- Lim, W.A., Richards, F.M., and Fox, R.O. 1994. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372 375–379. [DOI] [PubMed] [Google Scholar]
- Matthews, B.W. 1985. Determination of protein molecular weight, hydration, and packing from crystal density. Methods Enzymol. 114 176–187. [DOI] [PubMed] [Google Scholar]
- Mongiovi, A.M., Romano, P.R., Panni, S., Mendoza, M., Wong, W.T., Musacchio, A., Cesareni, G., and Fiore, P.P.D. 1999. A novel peptide-SH3 interaction. EMBO J. 18 5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A.J., Head, J.G., Barker, J.J., and Brady, R.L. 1998. Engineering an intertwined form of CD2 for stability and assembly. Nat. Struct. Biol. 5 778–782. [DOI] [PubMed] [Google Scholar]
- Nguyen, J.T., Turck, C.W., Cohen, F.E., Zuckermann, R.N., and Lim WA. 1998. Exploiting the basis of proline recognition by SH3 and WW domains: Design of N-substituted inhibitors. Science 282 2088–2092. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Owen, D.J., Wigge, P., Vallis, Y., Moore, J.D.A., Evans, P.R., and McMahon, H.T. 1998. Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J. 17 5273–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson, T. 1995. Protein modules and signaling networks. Nature 373 573–580. [DOI] [PubMed] [Google Scholar]
- Politou, A.S., Millevoi, S., Gautel, M., Kolmerer, B., and Pastore, A. 1998. SH3 in muscles: Solution structure of the SH3 domain from nebulin. J. Mol. Biol. 276 189–202. [DOI] [PubMed] [Google Scholar]
- Schlunegger, M.P., Bennett, M.J, and Eisenberg, D. 1997. Oligomer formation by 3D domain swapping: A model for protein assembly and misassembly. Adv. Protein Chem. 50 61–122. [DOI] [PubMed] [Google Scholar]
- Sicheri, F., Moarefi, I., and Kuriyan, J. 1997. Crystal structure of the Src family tyrosine kinase Hck. Nature 385 602–609. [DOI] [PubMed] [Google Scholar]
- Teeter, M.M. 1984. Water structure of a hydrophobic protein at atomic resolution: Pentagon rings of water molecules in crystals of crambin. Proc. Natl. Acad. Sci. 81 6014–6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanki, N., Thornton, J.M., Goodfellow, J.M. 1988. Distributions of water around amino acid residues in proteins. J. Mol. Biol. 202 637–657. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G, and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal, M., Montiel, J.L., Cussac, D., Cornille, F., Duchesne, M., Parker, F., Tocque, B., Roques, B.P., and Garbay, C. 1998. Differential interactions of the growth factor receptor-bound protein 2 N-SH3 domain with son of sevenless and dynamin. J. Biol. Chem. 273 5343–5348. [DOI] [PubMed] [Google Scholar]
- Wittekind, M., Mapelli, C., Farmer, II, B.T, Suen, K., Goldfarb, V., Tsao, J., Lavoie, T., Barbacid, M., Meyers, C.A., and Mueller, L. 1994. Orientation of peptide fragments from Sos proteins bound to the N-terminal SH3 domain of Grb2 determined by NMR spectroscopy. Biochemistry 33 13531–13539. [DOI] [PubMed] [Google Scholar]
- Wu, X., Knudsen, B., Feller, S.M., Zheng, J., Sali, A., Cowburn, D., Hanafusa, H., and Kuriyan, J. 1995. Structural basis for the specific interaction of lysine-containing proline-rich peptides with the N-terminal SH3 domain of c-Crk. Structure 3 215–226. [DOI] [PubMed] [Google Scholar]
- Xu, W., Harrison, S.C., and Eck, E.J. 1997. Three-dimensional structure of the tyrosine kinase c-Src. Nature 385 595–602. [DOI] [PubMed] [Google Scholar]