

Abstract
Carbonic anhydrases fall into three distinct evolutionary and structural classes: α, β, and γ. The β-class carbonic anhydrases (β-CAs) are widely distributed among higher plants, simple eukaryotes, eubacteria, and archaea. We have determined the crystal structure of ECCA, a β-CA from Escherichia coli, to a resolution of 2.0 Å. In agreement with the structure of the β-CA from the chloroplast of the red alga Porphyridium purpureum, the active-site zinc in ECCA is tetrahedrally coordinated by the side chains of four conserved residues. These results confirm the observation of a unique pattern of zinc ligation in at least some β-CAs. The absence of a water molecule in the inner coordination sphere is inconsistent with known mechanisms of CA activity. ECCA activity is highly pH-dependent in the physiological range, and its expression in yeast complements an oxygen-sensitive phenotype displayed by a β-CA-deletion strain. The structural and biochemical characterizations of ECCA presented here and the comparisons with other β-CA structures suggest that ECCA can adopt two distinct conformations displaying widely divergent catalytic rates.
Keywords: Carbonic anhydrase, crystal structure, metalloenzyme, zinc coordination, pH-dependent activity
The enzyme carbonic anhydrase (CA, carbonate hydrolase, EC 4.2.1.1) catalyzes a reaction of fundamental physiological and biochemical importance, the interconversion of the gaseous, membrane-permeable substrate carbon dioxide and the ionic compound bicarbonate as follows:
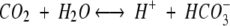 |
1 |
Recently, it has become clear that there are three phylogenetically and structurally distinct CA classes, designated α, β, and γ (Hewett-Emmett and Tashian 1996). Mammalian isozymes of the α class have been extensively characterized by physiological, structural, and mechanistic studies (for reviews, see Christianson and Cox, 1999 and Lindskog 1997). In contrast, structural and functional knowledge of the other two classes, β and γ, is much more limited. The recent discovery that these two classes are more broadly distributed among the domains of life has profound biological implications (Smith et al. 1999). Members of the β class, occurring mainly in higher plants, bacteria, and archaea, have in a few instances been shown as important and even essential components of metabolism in these organisms (Fukuzawa et al. 1992; Guilloton et al. 1993; Badger and Price 1994; Kozliak et al. 2000).
A common feature of all three CA classes is that they are zinc-containing metalloenzymes, suggesting that similar mechanistic principles apply to each. A zinc hydroxide mechanism for CO2 hydration, based principally upon studies of the paradigmatic α class enzyme, the human CAII isozyme (CAII; for a recent review, see Christianson and Cox 1999) has been well-established for this class. The essential elements of this mechanism, as represented by the following scheme
 |
2 |
are (a) nucleophilic attack of the metal-activated hydroxide ion on carbon dioxide, (b) ligand exchange of the product bicarbonate for a water molecule, and (c) regeneration of the zinc hydroxide form of the enzyme. The efficiency of step a rests upon the ability of zinc to act as a Lewis acid to stabilize a nucleophilic hydroxide ion, in effect lowering the pKa of H2O by up to seven or eight pH units (Christianson and Cox 1999). The ligand exchange step b may depend critically upon changes in metal coordination number or geometry. Step c requires intramolecular and intermolecular proton transfer from zinc-bound water ultimately to bulk solvent. These events, which are typically rate-limiting for the overall reaction, differ in detail between members of the α-class (Silverman and Lindskog 1988). Kinetic characterizations of β-CAs (Johansson and Forsman 1993; Rowlett et al. 1994) are consistent with a zinc hydroxide mechanism, whereas comparable studies of the only γ class enzyme to be characterized so far support a similar mechanism (Kisker et al. 1996; Alber et al. 1999).
Structural comparisons between the three classes reveal a diversity that is a remarkable counterpoint to the mechanistic similarities. Crystallographic analyses have been performed on γ-carbonic anhydrase from the methanogenic archaeon Methanosarcina thermophila (Kisker et al. 1996; Iverson et al. 2000) and the chloroplastic β-CAs from the photosynthetic red alga Porphyridium purpureum (Mitsuhashi et al. 2000) and Pisum sativum (garden pea; Kimber and Pai 2000). These studies have established the existence of three distinct molecular architectures for enzymes with CA activity. Despite the gross structural differences between the α and γ classes, both sequester the catalytic metal ion by the structurally superimposible imidazole groups of three histidine residues (Kisker et al. 1996). Furthermore, in the absence of bicarbonate or other zinc-binding small molecule, structures of both classes display additional coordination of the protein-bound zinc by water.
In sharp contrast to the α and γ class CAs, members of the β-CA class have significant α-helical character and employ a different set of zinc ligands. In both the crystal structure determined for the P. purpureum β-CA (PPCA; Mitsuhashi et al. 2000) and that for the P. sativum chloroplast β-CA (PSCA; Kimber and Pai 2000), zinc was found to be coordinated by two cysteines and one histidine residue. This was in agreement with predictions based on sequence homology among all β-CA sequences (Hewett-Emmett and Tashian 1996) and EXAFS and site-directed mutagenesis studies of pea and spinach chloroplasts β-CAs (Provart et al. 1993; Bracey et al. 1994; Rowlett et al. 1994). Additional features of the active sites observed in the PSCA structure, determined in the presence of the inhibitor acetate, were interpretable in terms of the zinc hydroxide mechanism. However, a very different active site is observed in the structure of PPCA. In this case, an aspartate residue, absolutely conserved in β-CAs, coordinates directly to zinc (Mitsuhashi et al. 2000). Missing from the tetrahedral coordination sphere is the catalytically activated water molecule common to uncomplexed α- and γ-CA structures, an observation difficult to reconcile with a zinc hydroxide mechanism.
Two members of the β class of carbonic anhydrases have been identified in E. coli. The first to be characterized was CynT, which is encoded by a part of the cyn operon (Sung and Fuchs 1988), whose function allows E. coli to utilize cyanate as a nitrogen source (Guilloton et al. 1992, 1993). The existence of a second β-CA homolog in E. coli was discovered as a result of efforts to sequence the E. coli genome (Fujita et al. 1994). Designated as cynT2 (Hewett-Emmett and Tashian 1996) or yadF (Fujita et al. 1994), the translated ORF displays 30% identity to CynT. Biochemical and functional information for the translation product of this gene has thus far been limited to that inferable by sequence homology alone.
The role of β-CAs in photosynthetic carbon fixation is supported by experimental evidence and theoretical arguments. The absence of CA activity in cyanobacteria and C4 higher plants is estimated to impair photosynthetic rates by a factor of about 104 (Badger and Price 1994). Less is known about the physiological roles performed by these enzymes in nonphotosynthetic organisms, but in general, loss of CA activity may result in growth defects. In some cases, such as acetogenic bacteria (Braus-Stromeyer et al. 1997) and methanogenic archaea (Smith and Ferry 1999), organisms that utilize CO2 in central metabolic processes (analogous to CO2 fixation in photosynthetic organisms), CA activity may be required to insure adequate levels of CO2 or HCO3− as a substrate for other enzymes. Genetic and physiological evidence indicates a requirement for the CynT β-CA to replenish bicarbonate in E. coli when cultures are grown at low pCO2 in the presence of cyanate (Guilloton et al. 1993; Kozliak et al. 2000). Direct evidence for β-CA activity in several pathogenic species of bacteria has been reported (Smith et al. 1999) and β-CA was identified in a screen for Salmonella typhimurium genes selectively induced upon infection of macrophages (Valdivia and Falkow 1997). The latter study also provided evidence that β-CA may play a role in virulence. In a competition assay, S. typhimurium strains with a targeted disruption of the β-CA gene showed decreased spleen colonization (Valdivia and Falkow 1997).
To contribute to a structure-based functional understanding of β carbonic anhydrases, we have chosen to investigate the mechanistic, functional, and regulatory properties of the E. coli carbonic anhydrase (ECCA) encoded by the cynT2/yadF gene.1 We have established that ECCA is an active carbonic anhydrase by in vitro assays and shown its ability to complement a growth-defect phenotype in β-CA-deficient yeast. We determined the crystal structure of ECCA to a resolution of 2.0 Å and have made detailed structural comparisons between ECCA and the PSCA and PPCA β-CAs. The pH-sensitivity observed for ECCA in vitro leads us to suspect that the activity of this oligomeric enzyme may be subject to regulation, whereas its correspondence with the zinc-ligation pattern observed for PPCA suggests the possibility that certain β-CAs may be capable of adopting either of two stable, alternate conformations, only one of which possesses significant CA activity.
Results
Variation of ECCA activity with pH
Although there is now evidence for the presence of active β-CAs in a large number of metabolically diverse prokaryotic species (Smith et al. 1999), further biochemical analysis of prokaryotic β-CA activity has been limited to the enzymes from the cyanobacterium Synechococcus PCC7942 (Fukuzawa et al. 1992), Cab; from the methanoarchaeon Methanobacterium thermoautotrophicum (Smith and Ferry 1999); and CynT from E. coli (Guilloton et al. 1992).
The ability of ECCA to catalyze the hydration of CO2 was determined using a colorimetric assay (Khalifah 1971) at several different starting pH values. These measurements indicate that ECCA is an active carbonic anhydrase at pH 8.4 (Fig. 1A ▶) and pH 9.1 (data not shown), but under the conditions used in our assay, no activity was detectable at pH 7.5 (Fig. 1B ▶). A zinc hydroxide mechanism for β-CAs implies that the rate of CO2 hydration will increase with increasing pH, because the proportion of the active, Zn2+−OH− form increases. Whereas other β-CAs show maximal activity at pH values near 9.0 under various conditions, their activity is significant at neutral and lower pH values. For example, the β-CA from spinach chloroplast has a kcat of 7.5 × 104 sec−1 at pH 7.0 (Rowlett et al. 1994) and the corresponding value for PSCA is 1.2 × 105 sec−1 (Johansson and Forsman 1993). Our data for ECCA suggest a steep variation in rate with pH, with the enzyme becoming inactive at neutral pH.
Fig. 1.


ECCA enzymatic assay progress curves for different pH values. Carbonic anhydrase (CA) activity was measured by an adaptation of the procedure of Khalifah (1971). (A) The data at pH 8.4 shows that the nonenzymatic hydration reaction approaches equilibrium within ∼15 sec under these conditions, whereas the progress curve for the sample containing ECCA (2.5 μM) overlaps that for the much more rapid bovine CA II positive control (α-CA). These data establish the ability of ECCA to catalyze the conversion of carbon dioxide to bicarbonate. (B) The same assay at pH 7.5 yields an ECCA progress curve that overlaps that of the nonenzymatic sample.
ECCA complements the growth defect in a yeast β-CA knockout
Because of the lack of any previous experimental data bearing on the in vivo role of this enzyme, we tested the ability of ECCA to complement a defect in a S. cerevisiae strain due to deletion of its homologous β-CA gene, NCE103. The deletion strain, ΔNCE103, displays an oxygen-sensitive growth-defect phenotype and in addition shows increased sensitivity to oxidative stress (Götz et al. 1999). We independently generated a ΔNCE103 strain for this investigation and created a yeast expression vector carrying the cynT2/yadF coding sequence. Although the ΔNCE103 strain is unable to grow under normal atmospheric (aerobic) conditions (Fig. 2 ▶), it is able to propagate in a low-oxygen (<0.1%) environment or in still-liquid cultures (data not shown). In contrast, as shown in Figure 2 ▶, the ΔNCE103 strain transformed with the ECCA expression vector (pMRC12) regains the ability to grow in an aerobic environment. The possible role of carbonic anhydrase in protection of cells from oxidative stress is not unprecedented (Räisänen et al. 1999), but the mechanism(s) for such protection in yeast remains to be elucidated. The possibility that ECCA performs a similar function in its native context awaits confirmation by a similar genetic approach in E. coli.
Fig. 2.

In an in vivo assay for ECCA function, a complementation experiment was performed in the yeast Saccharomyces cerevisiae. A mutant strain in which the yeast β-CA gene, NCE103, is inactivated by insertional mutagenesis (see Materials and Methods) displays inability to grow on solid media under normal, aerobic conditions (top right and bottom), in contrast to the wild type (w303–1a strain, top and bottom left). Complementation of the ΔNCE103 growth defect using a multicopy expression plasmid, pMRC12, encoding the cynT2/yadF ORF restores normal growth (bottom right). The asterisk denotes transformed strains cured of plasmid by treatment with 5-FOA, which provides an additional check that the rescued phenotype is conferred by pMRC12.
Structure determination and quality of the model
The crystal structure of ECCA has been determined in two different tetragonal crystal forms. Both crystal lattices reveal a tetrameric arrangement of ECCA monomers. The initial model for ECCA was obtained by MAD phasing using data from a crystal form (form 2) with two monomers per asymmetric unit (see Materials and Methods). Our discussion is mainly based on the form 1 structure, with one monomer per asymmetric unit, which has been solved to higher resolution (2.0 Å) with lower crystallographic R factor. The final model, a stereoview of which is presented in Figure 3A ▶, has been refined to an R factor of 17.7% and R free of 20.3% (Table 1). The RMSD value for main-chain atoms of a superposition of the three independent monomers is 0.56 Å.
Fig. 3.



(A) Stereoview of the Cα trace of the ECCA monomer, with every tenth position represented as a sphere and numbered. The N and C termini of the monomer are also indicated. Coloring is according to the subdomain structure of ECCA (see text), with the N-terminal arm in red, the zinc-binding core in yellow, and the C-terminal subdomain in blue. (B) Ribbon diagram of ECCA monomer, in approximately the same orientation as shown in A, with secondary structure elements labeled. Helices are shown in red, β-strands in yellow, and connecting segments are blue. The magenta sphere denotes the location of the zinc ion. C Structure-based sequence alignment of ECCA with the two other β-CAs for which crystal structures have been determined, the PPCA and the PSCA. PPCA actually contains two copies of the structural motif corresponding to the ECCA monomer that are ∼70% identical (Mitsuhashi et al. 2000). These are represented separately in the alignment as N and C. Residues that are identical in all four β-CA domains are boxed in red; those that are highly conserved are blue. Positions displaying conservative substitutions are shown in gray. Asterisks denote the residues observed as zinc ligands in the ECCA structure. The secondary structural features of ECCA are indicated above the alignment (helices indicated as cylinders, strands as arrows) and are colored according to the sequential ECCA subdomains, as shown in A. Due to the structural divergence among the three proteins in the region corresponding to the C terminus of ECCA (residues 196–220, which includes helix H10), these residues are excluded from this alignment.
Table 1.
Refinement statistics
| Form 1 | Form 2 | |
| Space group | P42212 | P4322 |
| Unit cell dimensions (Å) | a=b=68.54, c=85.88 | a=b=81.24, c=162.14 |
| Resolution (Å) | 2.0 | 2.2 |
| Figure of merit | 0.685 | — |
| Protein atoms | 1716 | 3353 |
| Water molecules | 62 | 87 |
| R factor/R free (%) | 17.7/20.3 | 21.6/24.7 |
| Reflections in R free set | 1379 | 2848 |
| RMS Δ bond lengths (Å) | 0.004 | 0.006 |
| RMS Δ bond angles (°) | 1.081 | 1.118 |
| Mean coordinate error (Å) | 0.20 | 0.27 |
Monomer fold
The general architecture of the ECCA monomer (Fig. 3 ▶) and secondary structural content place it in the α/β category (Levitt and Chothia 1976). A central four-stranded parallel β-sheet is flanked on one side by five major α-helical segments. The ECCA monomer is composed of three sequential components: an N-terminal arm, a conserved zinc-binding core, and a C-terminal subdomain that is dominated by α-helix (Fig. 3A,B ▶). The N-terminal arm is composed of two α-helical segments (H1 and H2) that extend away from the rest of the molecule and make significant contacts with an adjacent monomer. The following 70 residues (34–102) comprise a conserved and compact element of structure associated with zinc binding. In sequence alignments of the family of β-CA proteins, the highest degree of conservation occurs within this region, which includes residues that directly coordinate zinc or that lie within the immediate vicinity of the zinc-binding site (Fig. 3C ▶). This conserved zinc-binding core, which resembles the Rossmann fold nucleotide-binding motif, is composed of three of the four β strands (S1-S3) and the connecting segments between them (including helices H3 and H4), which pack against either side of the sheet. The residues observed coordinated to zinc in the ECCA structure—Cys42, Asp44, His98, and Cys101—occur at the end of or immediately following the β strands S1 and S3 (Fig. 3C ▶). Packed together in the C-terminal subdomain are six α-helices, two (H8 and H10) arranged antiparallel to each other and connected by a structure whose main chain forms a long, bent β hairpin, part of which forms the fourth strand of the parallel sheet.
Comparison of the ECCA monomer with these two previously determined β-CA structures shows significant differences in the C-terminal subdomain. Whereas in both the ECCA and PPCA structures, the C terminus forms an α-helix that packs against the rest of the monomer; in the PSCA structure, this section is in an extended conformation that projects outward from the monomer. As discussed below, the N-terminal extension and zinc-binding core mediate formation of a dimer in ECCA. This underlying dimeric association of monomers appears to be common to all three β-CA structures determined so far, whereas variation in the C-terminal subdomain contributes to the differing higher-order oligomerization observed for the PSCA enzyme.
Dimer structure
The MAD-phased ECCA structure was obtained from form 2 crystals that contain a dimer in the crystallographic asymmetric unit. The monomers composing the dimer are oriented in an antiparallel fashion, related by a twofold noncrystallographic symmetry axis (Fig. 4A ▶). The dimerization, which buries 2834 Å2 surface area per monomer, brings the zinc-binding cores of the adjacent monomers into close association, whereas the N-terminal arms reach across to contact portions of both the zinc-binding core and the C-terminal subdomain of the opposing monomer. The orientation and spacing of the two N-terminal arms in the dimer create the sides of a broad and deep groove in the dimer structure, reminiscent of a saddle (Fig. 4B ▶). The juxtaposition of the S2 strands of each monomer, which due to their crossing angle of 120° do not make hydrogen bonds through their main-chain groups, results in the formation of a pseudocontinuous eight-stranded β-sheet running through the core of the dimer. In the ECCA dimer, Ser 43, a well-conserved residue that is between two zinc-binding residues, makes contacts to S2 of the adjacent monomer. Residues from S2 (notably Phe 61), H4 (notably Tyr 83 and Leu 88), and the loop-connecting H2 and S1 make the closest approaches to the zinc associated with the opposing monomer. In summary, although zinc binding is wholly mediated by residues within a given monomer, interactions at the dimer interface contribute to the local environment at the metal-binding site.
Fig. 4.


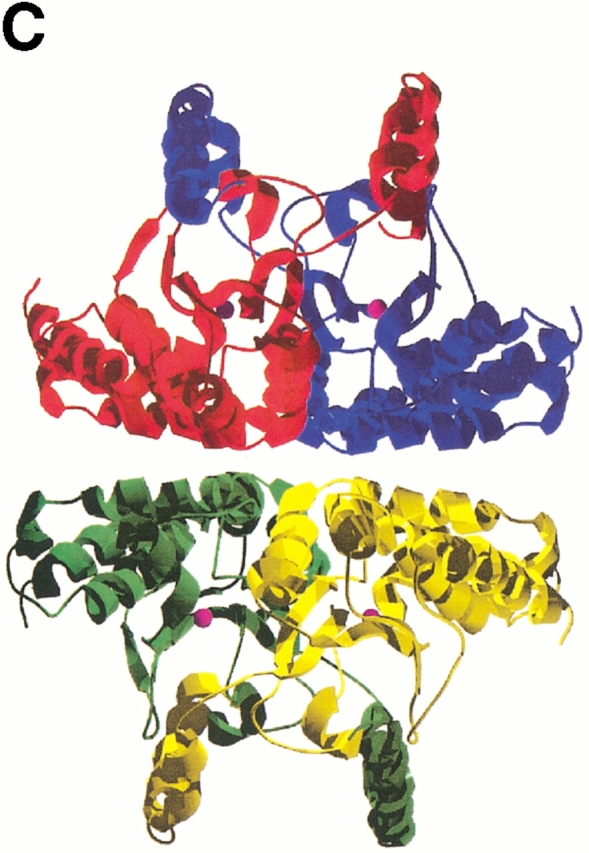
Ribbon representations of oligomeric associations of the ECCA monomer. (A) The dimer is shown, with each monomer colored by subdomain. The N-terminal arm is shown in red, the zinc-binding core in yellow, and the C-terminal subdomain in blue. In the second monomer, the respective colorings are magenta, green, and cyan. Magenta spheres represent the zinc atoms. (B) View of dimer rotated by 90° from that in A with respect to a horizontal axis through the center of the dimer. This view displays the saddle formed by the N-terminal arms, the proximity of the β-hairpin from the C-terminal subdomain to the latter, and the large twist of the central parallel β-sheet. (C) Tetrameric association observed in ECCA crystal lattices. Two dimers, the bottom one oriented as shown in B, associate along two perpendicular crystallographic twofold axes, forming a tetramer with 222 point symmetry. Each monomer is represented as a different colored ribbon, and the zinc atoms are shown as magenta spheres.
Tetramer formation
The dimer of the MAD-phased ECCA structure forms a close association with a symmetry-related dimer, suggesting that ECCA is a tetramer in solution (Fig. 4C ▶). This is consistent with the results of size-exclusion chromatography (data not shown) and is also in accord with the native molecular weights reported for other prokaryotic members of this family such as E. coli CynT (Guilloton et al. 1992) and M. thermoautotrophicum Cab (Smith and Ferry 1999), as well as that for PPCA (Mitsuhashi et al. 2000). This tetramer interface is less closely packed than the dimer interface, burying only 1848 Å2 of each dimer, about one-third the amount per monomer, as does dimerization. There are three main regions of contact between monomers across the tetramer interface that provide van der Waals and hydrogen-bond interactions: the loop between S2 and H4 in the zinc-binding core and along helices H6 (N-terminal portion) and H9 in the C-terminal subdomain. Interestingly, although PPCA displays an oligomeric association similar to ECCA, the presence of insertions in the PSCA sequence relative to ECCA (one just before H4 and another between H5 and H6, Fig. 3C ▶) seems to preclude this mode of association between dimers. These insertions correspond to extended loops that project across the ECCA tetramer interface. Alternatively, the PSCA dimers form two distinct associations mediated by the extended C-terminal tail, leading to a novel octameric arrangement (Kimber and Pai 2000).
Zinc-binding and active-site architecture
The crystal structure of ECCA shows nearly the same coordination to zinc, as was observed in the structure of β-CA from PPCA (Mitsuhashi et al. 2000). The carboxylate oxygen of Asp 44 provides a fourth-protein-derived ligand, in addition to Sγ atoms from Cys 42 and Cys 101 and Nδ1 of His 98. These four residues coordinate in a roughly tetrahedral arrangement (see Table 2 for a summary of the geometric parameters of zinc ligation), with Oδ2 of Asp 44 making the closest approach to the metal and water absent from the coordination sphere. This feature of the ECCA structure is quite clear in the experimental, MAD-phased electron density map obtained for two independent zinc sites (Fig. 5 ▶). The Asp 44 coordination displays a monodentate anti stereochemistry rarely observed in proteins (Christianson 1991) and the Oδ2-zinc bond lies 19° out of the plane of the carboxylate group. The positions of the cysteine Sγ atoms refine to coordination distances in excellent agreement with the range reported for Sγ−Zn2+ distances in small-molecule structures containing metal-thiolate complexes (Harding 1999) and the stereochemical features of the observed cysteine-zinc interactions conform to those typically observed for cysteine metal binding in proteins (Chakrabarti 1989). The Cys-Asp-His-Cys zinc-coordination scheme observed in both ECCA and PPCA is unique among zinc proteins in a metalloprotein structural database (Metalloprotein Structure and Design Group 1999).
Table 2.
Zinc coordination geometry
| Bond angles, X–Zn2+–Y (°) | ||||
| Distance to Zn2+ (Å) | Asp 44 Oδ2 | His 98 Nɛ2 | Cys 101 Sγ | |
| Cys 42 Sγ | 2.33 | 100.0 | 121.5 | 113.2 |
| Asp 44 Oδ2 | 2.10 | — | 100.3 | 112.3 |
| His 98 Nɛ2 | 2.21 | — | 108.3 | |
| Cys 101 Sγ | 2.37 | — | ||
| Bond angle (°) | Torsion angle (°) | |||
| Cys 42 | Cβ–Sγ–Zn2+ | 101.3 | Cα–Cβ–Sγ–Zn2+ | ∼180° |
| Asp 44 | Cβ–Oδ2–Zn2+ | 128.2 | ||
| His 98 | Cɛ1–Nɛ2–Zn2+ | 127.5 | ||
| His 98 | Cδ2–Nɛ2–Zn2+ | 120.4 | ||
| Cys 101 | Cβ–Sγ–Zn2+ | 103.1 | Cα–Cβ–Sγ–Zn2+ | ∼80° |
Fig. 5.
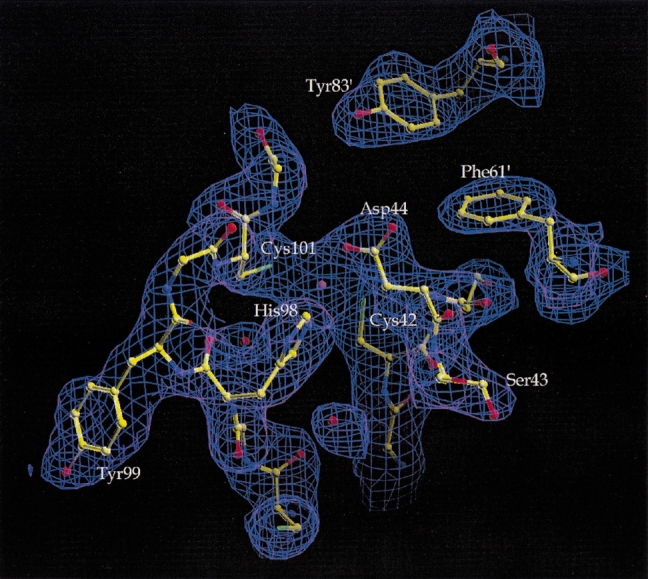
Electron density observed in the vicinity of the zinc cation. The MAD-phased map, following density modification, is shown contoured at 1.0 σ in blue. Superimposed upon the map is the final model of the form 2 crystal structure, represented in ball-and-stick form, with atoms and their bonds colored according to type: carbon (yellow), nitrogen (blue), oxygen (red), and sulfur (green). The zinc atom in the center of the figure is shown as a violet sphere; red spheres indicate water molecules. Note that the experimental density for the Asp44 side chain in anti monodentate coordination to zinc is clear and unequivocal.
Discussion
Comparative analysis of β-CA active sites
The distinctive contribution of the conserved aspartate to the zinc-coordination sphere is common to both the ECCA and PPCA crystal structures, but the latter differs in containing two distinct conformations of this aspartate and the following serine residue. One conformation, that of the C-terminal β-CA motif, corresponds to that observed in ECCA, which shows Asp 44 (Asp 405 in PPCA) in α-helical main-chain conformation and anti coordination to zinc, whereas Ser 45 (Ser 406 in PPCA) adopts a left-handed helical main-chain conformation. The N-terminal β-CA motif of PPCA shows the residue analogous to Asp 44 (Asp 151 in PPCA) with a main-chain φ angle between α and β conformations and zinc-coordination geometry closer to monodentate syn. The following serine residue adopts β main-chain angles. The significantly higher temperature factors for the main-chain atoms of these residues reported for PPCA (Mitsuhashi et al. 2000) are not mirrored in the ECCA structure—the average B is less than 20 Å2 for the eight main-chain atoms.
A significant contrast to the active sites of ECCA and PPCA was provided by the PSCA structure in the presence of acetate. Comparison of ECCA and PSCA structures reveals a pattern of differences that dramatically alter the zinc-coordination sphere and stereochemical organization of the active site (Fig. 6A ▶). In the uncomplexed form of the enzyme (as represented by the ECCA structure), one carboxylate oxygen of Asp 44 is able to participate in the coordination of zinc, whereas the other forms hydrogen bonds to two water molecules. In PSCA, acetic acid has replaced the Asp (Asp 162 in PSCA), which adopts a different side-chain rotamer and main-chain φ angle. The latter corresponds with a large main-chain displacement at Ser 45, Arg 46, and following residues in the superposition of ECCA and PSCA (Fig. 6B ▶). The left-handed helical conformation adopted by Ser 45 results in a hydrogen bond between its amide nitrogen and the carbonyl of Cys 42 (Fig. 6A ▶) that is absent in PSCA. On the other hand, PSCA shows a pair of hydrogen bonds between the residues corresponding to Asp 44 and Arg 46 not present in the ECCA structure. Other changes in the vicinity of the active site involve Arg 64 and Gln 33′. The latter residue is contributed by the opposing monomer, and the side chain of the corresponding residue in PSCA (Gln 151) is in position to form hydrogen bonds to an oxygen of the product-analog acetic acid and the hydroxyl from Ser 45.
Fig. 6.


(A) Stereoview of overlay of ECCA with PSCA showing conformational differences in the vicinity of the active site. The ECCA model is shown in stick form with atoms colored according to type: carbon (gray), nitrogen (blue), oxygen (red), and sulfur (yellow). Red spheres indicate waters, and the magenta sphere is the zinc (Zn2+) from the ECCA form 1 crystal structure. For the PSCA structure (1EKJ, monomers G and H), one monomer (G) is shown in dark green, and the juxtaposed monomer (H) of the dimer is in yellow; zinc is represented as a green sphere. The acetic acid molecule (HOAc) in the active site of the PSCA structure is shown colored according to atom type, as for the ECCA model. Some residues and water molecules have been omitted for clarity. The difference in side-chain rotamer and peptide flip of Asp44 is apparent, which results in a large main-chain displacement at Ser 45. The Ser 45 side chain is radically redirected away from the interaction seen in PSCA with the residue equivalent to Gln 33′. Reorientation of the side chain of Arg 64 appears correlated with the large differences at Ser 45. Other significant differences involve the positions of the Arg 46 and Gln 33′ side chains. Note that in ECCA, water molecules occupy positions close to those of side chains in PSCA. (B) Shift plot of main-chain residue root-mean square difference (RMSD) values for a superposition of the PPCA (1DDZ, N-terminal β-CA motif, red) and PSCA (green) structures upon the ECCA structure. The plot shows the calculated RMSD values for the main-chain atoms of each superimposed residue corresponding to ECCA residues 25–105. Displacements within the N-terminal arm (up to residue 33) are significantly greater than those within the zinc-binding core (residues 34–102). The peak for PSCA at position 45 reflects conformational differences that may correspond to switching between enzymatically active and inactive states (see text). The large peak for the PSCA structure centered at residue 73 corresponds to an insertion that projects across the tetramer interface of ECCA.
Another conformational difference involves the zinc-binding residue His 98. In the pea enzyme structure, the Nδ1 atom of the corresponding His 220 donates a hydrogen bond to the carbonyl oxygen of the following residue. The corresponding interaction is absent in both the ECCA and PPCA structures and a water molecule is within hydrogen-bonding distance of both atoms. The arrangement observed in ECCA and PPCA may relate to the tyrosine residue that follows histidine in both proteins, in contrast to PSCA and most β-CA sequences. This residue, Tyr 99 of ECCA, projects into a pocket of the C-terminal subdomain, near residues Glu 148 and Arg 189, and patterns of hydrogen bonding involving these residues can possibly impact the positioning of His 98.
Mechanistic and regulatory features of ECCA and β-CAs
Flexibility in coordination is a key feature of many zinc-enzyme mechanisms (Lipscomb and Sträter 1996). Structures of enzyme-bicarbonate complexes showing bicarbonate bound to zinc in a bidentate manner in both α- and γ-class CAs (Xue et al. 1993; Iverson et al. 2000) may represent alternate binding modes of the anionic product important in ligand exchange (step b of equation 2) and have been interpreted as evidence that, in the case of α-CAs, zinc undergoes a change of coordination from four to five ligands during catalytic turnover (Liljas et al. 1994). The distinct coordination states observed among the three βCA structures suggest that at least some β-CAs have the capacity to alter their zinc coordination in a manner implying the existence of either regulatory or mechanistic features that are novel in CA and zinc enzymology. In addition, although the precise pathways of proton transfer are not yet clearly defined for β-CAs, the structural information has identified residues (such as Tyr 83) with potential to function in such a role and clearly indicates that this rate-limiting process must differ in detail from that of the α class (Kimber and Pai 2000; Mitsuhashi et al. 2000).
In the ECCA crystal structure, the lack of closely coordinated water at the zinc cation (the closest water molecule is 4.3 Å from Zn2+) may correlate with the failure to detect ECCA carbonic anhydrase activity in assays at neutral pH (Fig. 1B ▶). The coordination of water to zinc would seem to be a prerequisite for the metal to act as a Lewis acid in the zinc hydroxide mechanism of CAs. Furthermore, the kinetic data so far obtained for β-CAs is consistent with the zinc hydroxide mechanism and provides no evidence for an alternative mechanism for the CA reaction. Thus, the mechanistic implications of the absence of water in the inner coordination sphere in the crystal structures of ECCA and PPCA (Mitsuhashi et al. 2000) must be carefully considered. Mitsuhashi et al. (2000) propose that Asp 44 (ECCA numbering) acts as a base to abstract a proton from a water molecule observed close to the second carboxylate oxygen of this residue (the first being coordinated to zinc). Asp 44 is then postulated to undergo a conformational change along with ligand exchange, allowing coordination of hydroxide ion to zinc and maintaining tetra coordination. On the other hand, the different position observed for the Asp 44 analog in the PSCA structure leads the investigators to predict quite a different role (Kimber and Pai 2000). Our comparison of these structures to each other and to that of ECCA suggests the possibility that conformational changes including, but not limited to, Asp 44 are associated with interconversion between enzymatically inactive and active forms.
One model for this interconversion is a concerted one in which conformational changes at Asp 44, Ser 45, and Arg 46 and a switch at Arg 64 are coupled to the reorganization of residues in the N-terminal arm near the active site. The coordination of zinc by Asp 44 precludes carbonic anhydrase activity; yet mutagenesis studies of β-CAs support an essential role for Asp 44 in catalysis (Bracey et al. 1994). Relief of this inhibition, as well as positioning of Asp 44 for a catalytic role, occurs by means of its interaction with Arg 46. At the same time Arg 46 and Ser 45 assist in the recruitment of the side chain of Gln 33′ to the active site. Moreover, the location of the Arg 64 guanidinium group is coupled to the main-chain displacements following Asp 44 through its hydrogen bond to the critical Asp 44 carbonyl in the PSCA conformation. This hypothesis provides a rationale for a critical role of Arg 46, which is the only residue other than those providing the four direct zinc ligands absolutely conserved in all known β-CA sequences. A specific prediction according to this model would be that a substitution of Arg 46 would severely compromise if not abolish CA activity mainly due to the increased propensity of Asp 44 to remain coordinated to zinc.
Although the existence of two stable conformations or states with widely differing activities has yet to be established for any β-CA, this hypothesis would also explain the apparent contradiction between lack of a zinc-coordinated water in the uncomplexed β-CA structures and clear kinetic evidence for a zinc hydroxide mechanism. The conformational switch producing movement of Asp 44 would be followed by coordination of water to zinc and an initial proton transfer event producing hydroxide ion. Persistence of an active conformation would allow continuous turnover according to the mechanism proposed by Kimber and Pai (2000). Interestingly, a dependence of CA activity upon pH similar to that observed for ECCA was reported for PPCA (Yagawa et al. 1987), supporting the idea that if two stable states of β-CA can exist in solution, the active form may be favored by high pH.
We cannot rule out the possibility that conformational change coupled to ligand exchange involving Asp 44 occurs repeatedly within continuous catalytic turnover. In such a scenario, the rapid conformational changes might allow Asp 44 to play a role in proton transfer and facilitating product release (Mitsuhashi et al. 2000). Of relevance in this regard, are two examples of other zinc enzymes in which structural evidence indicates displacement of a protein ligand to zinc during catalysis. In the crystal structure of unliganded alkaline protease from Pseudomonas aeruginosa, the catalytic zinc is observed to be ligated by a tyrosine residue (Miyatake et al. 1995), whereas in the form of the enzyme with bound heterogeneous tetrapeptide, this tyrosine is displaced, forming a hydrogen bond to one of the carboxylate oxygens of the C terminus of the tetrapeptide (Baumann et al. 1993). L-fuculose-1-phosphate aldolase from E. coli crystallizes in an unliganded form with zinc coordinated in a bidentate manner by a glutamate (Glu 73) residue (Dreyer and Schulz 1993). The structure of the same enzyme in the presence of the inhibitor phosphoglycolohydroxamate revealed a rotation of the Glu 73 side chain into a less polar environment in which one of the carboxylate oxygens forms a hydrogen bond to a main-chain amide group. The other carboxylate oxygen is postulated to play a role as a proton acceptor in a model for the enzymatic mechanism (Dreyer and Schulz 1996). In both examples, the coordination change and other conformational differences appear to be substrate induced and the catalytic rates are quite modest. Fuculose-1-phosphate aldolase has a turnover number of 19 sec−1 (Joerger et al. 2000). The slow rates of these enzymes and the fact that the substrate of the CA reaction in the forward direction, namely water, is freely available to the ECCA protein within the environment of the crystal lattice, favor the possibility that the conformation at the β-CA active site observed for ECCA and PPCA represents a stable, inactive form perhaps reflecting a regulatory process.
The β-CA active site may represent a striking example of the critical role of flexibility of coordination geometry in metalloenzymes. An inherent flexibility in coordination is strongly implied by the zinc coordination observed in the ECCA and PPCA structures. The structure of a mutant of human CAII in which Thr 199 is substituted with glutamate shows the latter coordinated as a fourth-protein ligand to zinc with geometry similar to that of ECCA Asp 44 (Ippolito et al. 1995). The catalytic activity of the mutant, however, was nearly obliterated, suggesting that flexibility permitting coordination of water, as required for the zinc hydroxide mechanism, could not be accommodated within the framework of the α-type CAII structure. Although dynamic coordination behavior is a common characteristic of catalytic zinc (Christianson 1991), our analysis raises the possibility that in the case of some β-CAs, the ability to adopt alternate coordination states is employed as a regulatory mechanism.
Materials and methods
Protein purification, crystallization, and diffraction data acquisition
The cloning, expression, and purification of ECCA (Cyn T2), as well as crystallization and data-collection methods, have been described in more detail elsewhere (Cronk et al. 2000). Briefly, the coding sequence for ECCA was amplified from E. coli DNA using PCR and then the product was cloned into a pET-3a expression vector (Novagen). The overexpressed protein was obtained from transformed E. coli cultures and purified by metal (Ni2+) affinity chromatography. Crystallizations were performed by vapor diffusion from hanging drops that were mixtures of 2–3 μL protein at 12 mg/mL in TBS (20 mM Tris-HCl, 100 mM NaCl at pH 8.3) with an equivalent volume of reservoir solution of the compositions indicated below and were equilibrated against 1 mL of reservoir solution. Crystals of space group P42212 (form 1) grow using 1.8–1.6 M ammonium sulfate and 0.1 M MES at pH 6.3 at 20°C. Form 2 crystals (space group P4322) form upon addition of 4% PEG 400 to the form 1 conditions. Data for form 1 crystals were collected in-house at room temperature using a rotating anode generator and image plate detector. The multiwavelength anomalous dispersion data for form 2 crystals were acquired at 100 K on beamline 5.0.2 of the Macromolecular Crystallography Facility at the Advanced Light Source (Lawrence Berkeley National Laboratory). The ALS data set was collected at four wavelengths at or near the Se edge in the vicinity of 0.98 Å, plus a fifth wavelength, 1.28 Å, corresponding to the peak anomalous scattering for zinc.
The acquired data were reduced and scaled in preparation for phase calculations using Elves (J. Holton and T. Alber, unpubl.), which administered intensity measurements using MOSFLM (Leslie 1992) and data-scaling procedures by SCALA (CCP4, 1994). The resulting scaled and merged structure factor amplitudes were analyzed for anomalous and dispersive difference signals using the XtalView program package (McRee 1992) and CNS (Brünger et al. 1998).
Structure determination by MAD phasing (form 2)
An anomalous Patterson map calculated using data collected at 1.28 Å (corresponding to the peak anomalous scattering signal for zinc) was clearly interpretable as two Zn2+ sites in space group P4322 (P4122). The sites found manually were refined by Xheavy (McRee 1992) and used to calculate phases for an anomalous difference Fourier map, combined with amplitudes from the data from the Se anomalous peak. The top six peaks in this map corresponded with one of the solutions produced by the CNS heavy search program (Brünger et al. 1998) from the input Se anomalous data. The positions of the two Zn and six Se in the asymmetric unit were refined against data from all five wavelengths and then used to calculate MAD phases to 3.0 Å, which led to a figure of merit of 0.685. The MAD-phased electron density was subjected to density modification in CNS (Brünger et al. 1998), including density truncation, solvent flipping (solvent content 54%), and phase extension to 2.25 Å. The resulting map was clearly interpretable, and a C-alpha trace was made using the program Xfit (McRee 1992). A nearly complete model was built according to the tracing within the program O (Jones et al., 1991) with manual adjustments to improve the fit of the model to the experimentally observed electron density.
Molecular replacement (form 1)
The structure of ECCA in the form 1 crystal was solved using molecular replacement with a model of the form 2 structure as a search model. The room temperature data set for the form 1 crystal, along with a monomer from the form 2 structural model at an early stage of refinement, were input to the molecular replacement program EPMR (Kissinger et al. 1999). The real-space correlation search was conducted in space group P42212. A solution with correlation coefficient 0.669 and R factor 39.1% was obtained in the first round of the search. This solution was used to calculate phases for a map using the observed form 1 structure factor amplitudes in CNS (Brünger et al. 1998). The correspondence of the model to the map and the crystal packing was examined in the molecular graphics program O (Jones et al. 1991).
Refinement (forms 1 and 2)
No NCS constraints were used in the map calculations or the model refinement of the form 2 crystal structure. The initial model contained residues 3–213 of each of the two monomers in the asymmetric unit. Several rounds of positional and B-factor refinement with TNT (Tronrud et al. 1987), followed by manual rebuilding of the model (including placement of two zinc atoms and water molecules), were performed. This yielded a model with good geometry and an R factor of 21.5% and R free of 29.5%. The model was then further refined by application of simulated annealing, torsional, and individual B-factor refinement with maximum likelihood targets using CNS (Brünger et al. 1998). Water molecules were placed and retained in the model by strict criteria of difference density, B-factor cutoffs, and hydrogen-bonding capacity. The R factor remained at 21.5%, whereas R free dropped to 24.5%. Models at several stages of the refinement were carefully evaluated by goodness of fit to simulated annealing composite omit maps calculated in CNS (Brünger et al. 1998). The final model includes residues 2–215 for monomer A and 3–215 for monomer B, which excludes several residues at the N termini and residues 216–220 at the C termini due to lack of interpretable electron density in these regions. The refinement of the form 1 structure was carried out by essentially the same procedure as for form 2, starting with the molecular-replacement solution. The final model includes residues 2–215 of the single monomer in the asymmetric unit.
Enzymatic activity measurements
Carbonic anhydrase activity was assayed by the changing pH/dye indicator method (Khalifah 1971). All assays were performed at room temperature. The buffer/indicator pairs used were 25 mM HEPES, 100μM phenol red (pH 7.5, λmax= 558 nm) and 25 mM TAPS, 100μM m-cresol purple (pH 8.4, λmax= 578 nm), and both solutions also contained 100 mM sodium sulfate to maintain ionic strength of the reaction medium. The reaction was initiated by addition of 0.5 ml CO2 (aq) to 0.5 mL of a 2× buffer indicator solution containing enzyme samples. ECCA activity was measured at a final concentration of 2.5 μM. A nonenzymatic control was included in each set of measurements and bovine carbonic anhydrase II (Sigma) was used as a positive control. Data were acquired at the appropriate wavelength at 0.1-sec intervals using a Varian Bio100 spectrophotometer with time T = 0 coinciding with manual addition of substrate.
Construction of yeast-deletion strain and complementation assay
The yeast-deletion strain was generated by homologous recombination upon transformation with linear dsDNA. PCR primers with 40 nt at their 5′ ends complementary to regions at the ends of the target NCE103 gene and 20 nt at their 3′ ends complementary to a regions flanking kanamycin resistance cassette in a pRS400 vector were synthesized. These primers were used with the pRS400 as template, yielding a 1.6-kb PCR product. The strain w303–1a was then transformed with the linear dsDNA PCR product and transformants were selected by plating onto C media containing the kanamycin analog G418 under low oxygen (<0.1%) conditions. The colonies obtained were analyzed by PCR for the presence of the replacement at the NCE103 locus, and the oxygen-sensitive phenotype was confirmed. Next, a yeast-expression plasmid, designated pMRC12, was constructed by ligation of the 660-bp CynT2 ORF with EcoRI linkers into EcoRI-digested and alkaline phosphatase–treated p426 GPD vector (Mumberg et al. 1995), containing a URA3 selectable marker. This expression construct was transformed into the knockout strain and transformants were selected on C minus URA media.
Accession numbers
The coordinates of both forms 1 and 2 crystal structure of ECCA have been deposited in the PDB database with accession codes 1I6P and 1I6O, respectively.
Acknowledgments
We thank M. Dubois, J. Holton, and D. Gottschling for technical advice; B. Stoddard, R. Strong, P. Anderson, R. Rowlett, and M. Kimber for illuminating discussions and critical readings of the manuscript; T. Alber and A. Ferre-D'Amare for use of facilities and equipment. J.D.C. and M.R.C. dedicate this work to the memory of Agnes "Nina" Hartman. This work was supported in part by grants from PHS NRSA T32 GM07270 from National Institutes of Health and from the Fred Hutchinson Cancer Research Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CA, carbonic anhydrase
ECCA, Escherichia coli β-carbonic anhydrase
PPCA, Porphyridium purpureum β-carbonic anhydrase
PSCA, Pisum sativum β-carbonic anhydrase
EXAFS, extended X-ray absorption fine structure spectroscopy
MAD, multiwavelength anomalous dispersion
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.46301.
Footnotes
The genetic nomenclature in this case is not particularly precise in conveying the relevant features of this transcriptional unit. The cynT2 designation is confusing and wrongly implies that the gene is part of the cyn operon. The yadF designation is not based on a functional characterization of the gene product as a carbonic anhydrase. Therefore, we will refer in this paper to the cynT2/yadF gene product simply as ECCA, for E. coli carbonic anhydrase.
References
- Alber, B.E., Colangelo, C.M., Dong, J., Stalhandske, C.M., Baird, T.T., Tu, C., Fierke, C.A., Silverman, D.N., Scott, R.A., and Ferry, J.G. 1999. Kinetic and spectroscopic characterization of the gamma-carbonic anhydrase from the methanoarchaeon Methanosarcina thermophila. Biochemistry 38 13119–13128. [DOI] [PubMed] [Google Scholar]
- Badger, M.R. and Price, G.D. 1994. The role of carbonic anhydrase in photosynthesis. Ann. Rev. Plant. Physiol. Plant Mol. Biol. 45 369–392. [Google Scholar]
- Baumann, U., Wu, S., Flaherty, K.M., and McKay, D.B. 1993. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: A two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 12 3357–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracey, M.H., Christiansen, J., Tovar, P., Cramer, S.P., and Bartlett, S.G. 1994. Spinach carbonic anhydrase: Investigation of the zinc-binding ligands by site-directed mutagenesis, elemental analysis, and EXAFS. Biochemistry 33 13126–13131. [DOI] [PubMed] [Google Scholar]
- Braus-Stromeyer, S.A., Schnappauf, G., Braus, G.H., Göβner, A.S., and Drake, H.R. 1997. Carbonic anhydrase in Acetobacterium woodii and other acetogenic bacteria. J. Bacteriol. 179 7197–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. 54 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4). 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, P. 1989. Geometry of interaction of metal ions with sulfur-containing ligands in protein structures. Biochemistry 28 6081–6085. [DOI] [PubMed] [Google Scholar]
- Christianson, D.W. 1991. Structural biology of zinc. Adv. Prot. Chem. 42 281–355. [DOI] [PubMed] [Google Scholar]
- Christianson, D.W. and Cox, J.D. 1999. Catalysis by metal-activated hydroxide in zinc and manganese metalloenzymes. Annu. Rev. Biochem. 68 33–57. [DOI] [PubMed] [Google Scholar]
- Cronk, J.D., O'Neill, J.W., Cronk, M.R., Endrizzi, J.A. and Zhang, K.Y.J. 2000. Crystallization and preliminary characterization of beta carbonic anhydrase from Escherichia coli. Acta Crystallogr. D. 52 1176–1179. [DOI] [PubMed] [Google Scholar]
- Dreyer, M.K., Schulz, G.E. 1993. The spatial structure of the class II l-fuculose-1-phosphate aldolase from Escherichia coli. J. Mol. Biol. 231 549–553. [DOI] [PubMed] [Google Scholar]
- Dreyer, M.K. and Schulz, G.E. 1996. Catalytic mechanism of the metal-dependent fuculose aldolase from Escherichia coli as derived from the structure. J. Mol. Biol. 259 458–466. [DOI] [PubMed] [Google Scholar]
- Fujita, N., Mori, H., Yura, T., and Ishihama, A. 1994. Systematic sequencing of the Escherichia coli genome: Analysis of the 2.4–4.1 min (110.917–193,643 bp) region. Nucleic Acids Res. 22 1637–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzawa, H., Suzuki, E., Komukai, Y., and Miyachi, S. 1992. A gene homologous to chloroplast carbonic anhydrase (icfA) is essential to photosynthetic carbon dioxide fixation by Synechococcus PCC7942. Proc. Natl. Acad. Sci. 89 4437–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz, R., Gnann, A., and Zimmerman, F.K. 1999. Deletion of the carbonic anhydrase-like gene NCE103 of the yeast Saccharomyces cerevisiae causes an oxygen-sensitive growth defect. Yeast 15855–864. [DOI] [PubMed] [Google Scholar]
- Guilloton, M.B., Korte, J.J., Lamblin, A.F., Fuchs, J.A., and Anderson, P.M. 1992. Carbonic anhydrase in Escherichia coli: A product of the cyn operon. J. Biol. Chem. 267 3731–3734. [PubMed] [Google Scholar]
- Guilloton, M.B., Lamblin, A.F., Kozliak, E.I., Gerami-Nejad, M., Tu, C., Silverman, D., Anderson, P.M., and Fuchs, J.A. 1993. A physiological role for cyanate-induced carbonic anhydrase in Escherichia coli. J. Bacteriol. 175 1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, M.M. 1999. The geometry of metal-ligand interactions relevant to proteins. Acta Crystallogr. D. 55 1432–1443. [DOI] [PubMed] [Google Scholar]
- Hewett-Emmett, D. and Tashian, R.E. 1996. Functional diversity, conservation and convergence in the evolution of α-, β-, and γ-carbonic anhydrase gene families. Mol. Phylogen. Evol. 5 50–77. [DOI] [PubMed] [Google Scholar]
- Ippolito, J.A., Baird T.T., McGee, S.A., Christianson D.W., and Fierke, C.A. 1995. Structure-assisted redesign of a protein-zinc binding site with femtomolar affinity. Proc. Natl. Acad. Sci. 92 5017–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson, T., Alber, B.E., Kisker, C., Ferry, J.G., and Rees, D.C. 2000. A closer look at the active site of γ-class carbonic anhydrases: High-resolution crystallographic studies of the carbonic anhydrase from Methanosarcina thermophila. Biochemistry 39 9222–9231. [DOI] [PubMed] [Google Scholar]
- Joerger, A.C., Gosse, C., Fessner, W-D., and Schulz, G.E. 2000. Catalytic action of fuculose 1-phosphate aldolase (class II) as derived from structure-directed mutagenesis. Biochemistry 39 6033–6041. [DOI] [PubMed] [Google Scholar]
- Johansson, I-M. and Forsman, C. 1993. Kinetic studies of pea carbonic anhydrase. Eur. J. Biochem. 218 439–446. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Khalifah, R.G. 1971. The carbon dioxide hydration activity of carbonic anhydrase. J. Biol. Chem. 246 2561–2573. [PubMed] [Google Scholar]
- Kimber, M.S. and Pai, E.F. 2000. The active site architecture of Pisum sativum β-carbonic anhydrase is a mirror image of that of α-carbonic anhydrases. EMBO J. 19 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisker, C., Schindelin, H., Alber, B.E., Ferry, J.G., and Rees, D.C. 1996. A left-handed β-helix revealed by the crystal structure of a carbonic anhydrase from the archaeon Methanosarcina thermophila. EMBO J. 15 2323–2330. [PMC free article] [PubMed] [Google Scholar]
- Kissinger, C.R., Gehlhaar, D.K., and Fogel, D.B. 1999. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr. D. 55 484–491. [DOI] [PubMed] [Google Scholar]
- Kozliak, E.I., Guilloton, M.B., Fuchs, J.A., and Anderson, P.M. 2000. Bacterial carbonic anhydrases. In The carbonic anhydrases: New horizons (eds. R.W. Chegwidden et al.), pp. 547–565. Birkhauser, Basel, Switzerland. [DOI] [PubMed]
- Leslie, A.G.W. 1992. Recent changes to the MOSFLM package for processing film and image data. Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography. Daresbury Laboratory, Warrington, UK.
- Levitt, M. and Chothia, C. 1976. Structural patterns in globular proteins. Nature 261 552–558. [DOI] [PubMed] [Google Scholar]
- Liljas, A., Håkansson, K., Jonsson, B.H., and Xue, Y. 1994. Inhibition and catalysis of carbonic anhydrase. Eur. J. Biochem. 219 1–10. [DOI] [PubMed] [Google Scholar]
- Lindskog, S. 1997. Structure and mechanism of carbonic anhydrase. Pharmacol. Ther. 74 1–20. [DOI] [PubMed] [Google Scholar]
- Lipscomb, W.N. and Sträter, N. 1996. Recent advances in zinc enzymology. Chem. Rev. 96 2375–2433. [DOI] [PubMed] [Google Scholar]
- McRee, D.E. 1992. A visual protein crystallographic software system for X11/XView. J. Mol. Graph. 10 44–46. [Google Scholar]
- Metalloprotein Structure and Design Group . 1999. Metalloprotein site database and browser (MDB). MDB, Release 1.4. http://metallo.scripps.edu/.
- Miyatake, H., Hata, Y., Fujii, Y., Hamada, K., Morihara, K., and Katsube, Y. 1995. Crystal structure of the unliganded alkaline protease from Pseudomonas aeruginosa IFO3080 and its conformational changes on ligand binding. J. Biochem. 118 474–479. [DOI] [PubMed] [Google Scholar]
- Mitsuhashi, S., Mizushima, T., Yamashita, E., Yamamoto, M., Kumasaka, T., Moriyama, H., Ueki, T., Miyachi, S., and Tsukihara, T. 2000. X-ray structure of β-carbonic anhydrase from the red alga, Porphyridium purpureum, reveals a novel catalytic site for CO2 hydration. J. Biol. Chem. 275 5521–5526. [DOI] [PubMed] [Google Scholar]
- Mumberg, D., Müller, R., and Funk, M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156 119–122. [DOI] [PubMed] [Google Scholar]
- Provart, N.J., Majeau, N., and Coleman, J.R. 1993. Characterization of pea chloroplastic carbonic anhydrase. Expression in Escherichia coli and site-directed mutagenesis. Plant Mol. Biol. 22 937–943. [DOI] [PubMed] [Google Scholar]
- Räisänen, S.R., Lehenkari, P., Tasanen, M., Rahkila, P., Härkönen, P.L., and Väänänen, H.K. 1999. Carbonic anhydrase III protects cells from hydrogen peroxide–induced apoptosis. FASEB J. 13 513–522. [DOI] [PubMed] [Google Scholar]
- Rowlett, R.S., Chance, M.R., Wirt, M.D., Sidelinger, D.E., Royal, J.R., Woodroffe, M., Wang, Y-FA., Saha, R.P., and Lam, M.G. 1994. Kinetic and structural characterization of spinach carbonic anhydrase. Biochemistry 33 13967–13976. [DOI] [PubMed] [Google Scholar]
- Silverman, D.N. and Lindskog, S. 1988. The catalytic mechanism of carbonic anhydrase: Implications of a rate-limiting protolysis of water. Acc. Chem. Res. 21 30–36. [Google Scholar]
- Smith, K.S. and Ferry, J.G. 1999. A plant-type (β-Class) carbonic anhydrase in the thermophilic methanoarchaeon Methanobacterium thermoautotrophicum. J. Bacteriol. 181 6247–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, K.S., Jakubzick, C., Whittam, T.S., and Ferry, J.G. 1999. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc. Natl. Acad. Sci. 96 15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, Y-c. and Fuchs, J.A. 1988. Characterization of the cyn operon in Escherichia coli K12. J. Biol. Chem. 263 14769–14775. [PubMed] [Google Scholar]
- Tronrud, D.E., TenEyck, L.F., and Matthews, B.W. 1987. TNT: TenEyck-Tronrud refinement package. Acta Crystallogr. A. 43 489–503. [Google Scholar]
- Valdivia, R.H. and Falkow, S. 1997. Fluorescence-based isolation of bacterial genes expressed within host cells. Science 277 2007–2011. [DOI] [PubMed] [Google Scholar]
- Xue, Y., Vidgren, J., Svensson, L.A., Liljas, A., Jonsson, B-H., and Lindskog, S. 1993. Crystallographic analysis of Thr-200 → His human carbonic anhydrase II and its complex with the substrate, HCO3–. Proteins 15 80–87. [DOI] [PubMed] [Google Scholar]
- Yagawa, Y., Muto, S., and Miyachi, S. 1987. Carbonic anhydrase of a unicellular red alga Porphyridium cruentum R-1. I. Purification and properties of the enzyme. Plant Cell Physiol. 28 1253–1262. [Google Scholar]