

Abstract
The endoprotease furin, which belongs to the family of mammalian proprotein convertase (PC), is synthesized as a zymogen with an N-terminal, 81-residue inhibitory prodomain. It has been shown that the proenzyme form of furin undergoes a multistep `autocatalytic' removal of the prodomain at the C-terminal side of the two consensus sites, R78-T-K-R81∼ and R44-G-V-T-K-R49∼. The furin-mediated cleavage at R44-G-V-T-K-R49∼, in particular, is significantly accelerated in an `acidic' environment. Here, we show that under neutral pH conditions, the inhibitory prodomain of furin is partially folded and undergoes conformational exchanges as indicated by extensive broadening of the NMR spectra. Presence of many ring-current shifted methyl resonances suggests that the partially folded state of the prodomain may still possess a `semirigid' protein core with specific packing interactions among amino acid side chains. Measurements of the hydrodynamic radii and compaction factors indicate that this partially folded state is significantly more compact than a random chain. The conformational stability of the prodomain appears to be pH sensitive, in that the prodomain undergoes an unfolding transition towards acidic conditions. Our NMR analyses establish that the acid-induced unfolding is mainly experienced by the residues from the C-terminal half of the prodomain (residues R44–R81) that contains the two furin cleavage sites. A 38-residue peptide fragment derived from the entire pH-sensitive C-terminal region (residues R44–R81) does not exhibit any exchange-induced line broadening and adopts flexible conformations. We propose that at neutral pH, the cleavage site R44-G-V-T-K-R49∼ is buried within the protein core that is formed in part by residues from the N-terminal region, and that the cleavage site becomes exposed under acidic conditions, leading to a facile cleavage by the furin enzyme.
Keywords: Prodomain, furin, heteronuclear NMR, protease activation, convertases
Most proteolytic enzymes are synthesized as zymogens with proregions that are typically N-terminal extensions of the mature enzymes (Khan and James 1998). The functions of the proregions appear to be multitudinous. Seminal studies have been performed with subtilisins and the α-lytic protease that show the ability of the isolated proregions to assist fast refolding of the denatured enzymes (Silen and Agard 1989; Shinde and Inouye 1993; Ruvinov et al. 1997). Proregions are often involved in the regulation of enzymatic activities of the cognate proteases by means of selective inhibition either in cis or in trans (Khan and James 1998). These studies indicate that the `prodomains' are separate functional modules of the protease zymogens. Apart from the chaperonin-like and inhibitory activities, proregions are found to be involved in targeting the proteases to specific organelles (Valls et al. 1990; Takeshima et al. 1995), membrane associations (McIntyre and Erickson 1993; Jan et al. 1998), and glycosylations (Chapman et al. 1997). Under in vivo conditions, temporal inhibition imparted by the proregions regulates premature proteolytic activities of the proteases that could lead to unwanted protein degradation. The `activation' of the proteases refers to the proteolytic cleavage of the inhibitory prosegments, leading to the release of the active enzymes. The cleavage of the proregions can be achieved by other enzymes intermolecularly or by the same enzyme in an intramolecular or autocatalytic pathway. It has been shown that the acidic environment is the most common parameter in triggering the activation of cysteine, aspartic acid, and metalloproteases zymogens (Erickson 1989; Takeda-Ezaki and Yamamoto 1993; Mach et al. 1994).
Mammalian PCs belong to the family of subtilisin-like serine endoproteases, which are responsible for the posttranslational processing of a variety of higher molecular weight precursor proteins within the secretory pathway (Van de Ven et al. 1993; Seidah et al. 1994; Seidah 1995; Nakayama 1997; Seidah and Chrétien 1997). The endoproteolytic activity of the convertases is manifested by the cleavage of protein substrates at specific paired residues or multiple basic residues (Nakayama 1997). At present, there are eight distinct convertases that are homologous with bacterial subtilisins and the yeast-processing enzyme kexin (kex2p) (Nakayama 1997; Siezen and Leunissen 1997; Seidah and Chrétien 1999). Furin was the first mammalian convertase discovered, and is the most extensively studied in the family (Roebroke et al. 1986; Fuller et al. 1989). Like many other proteases, furin is synthesized as an inactive precursor with an N-terminal, 81-residue proregion. Initially, it has been shown that the furin zymogen undergoes an autocatalytic multistep processing of its proregion, where the cleavage occurs at the C-terminal side of the two consensus-furin-recognition sites, R78-T-K-R81∼, and R44-G-V-T-K-R49∼. The second furin-mediated cleavage at R44-G-V-T-K-R49∼ of the prodomain is significantly accelerated in a slightly acidic environment (Anderson et al. 1997).
In this study, we show that the prodomain of furin is only partially folded under physiological conditions. Regardless, the prodomain undergoes a further unfolding in response to acidic pH, whereby the C-terminal region is more susceptible to pH-induced denaturation as compared with the N-terminal region. We surmise that the pH-induced unfolding transition of the furin prodomain could be related to the activation pathway of the furin zymogen, where the region encompassing the cleavage site R44-G-V-T-K-R49∼ is presumably hidden in a conformationally restricted environment by the residues from the N-terminal part of the prodomain.
Results
Conformational transitions in the furin prodomain
Figure 1 ▶ shows the low-field and up-field methyl resonances of the proton NMR spectra of the furin prodomain under neutral (pH 6.8) and acidic (pH 4.0) conditions. Proton spectra at the neutral pH are characterized by extensive broadening of almost all resonances as shown in Figure 1a ▶ for the aromatic ring protons, the amide protons, and the most downfield-shifted Nɛ1H resonances from the indole ring of the three tryptophan residues. The upfield-shifted methyl resonances are also considerably broadened. At a very low concentration (i.e., ∼0.02 mM of the prodomain) there was no apparent change in the line widths of all the proton resonances, precluding significant aggregation as the source of the broad resonances. The broad line widths observed at neutral pH for the prodomain are therefore a consequence of intramolecular conformational transitions between differently populated structures at microsecond-to-millisecond time scale (Wüthrich 1986). Even so, there are extremely upfield-shifted or "ring current" affected methyl resonances between 0.5 ppm and 0 ppm (Fig. 1a ▶, right panel), indicating tertiary packing interactions for at least some populations of the conformational ensemble. Observation of significantly downfield-shifted amide proton resonances at 9.0 ppm–9.2 ppm suggests the presence of hydrogen-bonded secondary structural elements (Fig. 1a ▶, left panel). These spectral features are also typically observed for the molten globule-like states of many proteins generated under mild denaturing conditions (Baum et al. 1989; Alexandrescu et al. 1993; Bhattacharjya and Balaram 1997; Eliezer et al. 1998).
Fig. 1.

One-dimensional 1H NMR spectra of the prodomain of furin at pH 6.8 (a) and pH 4.0 (b) in a solution of 50 mM sodium acetate. Regions shown are the low-field amide (7.8 ppm–9.2 ppm), aromatic (6.5 ppm–7.5 ppm) and the most down-field-shifted indole Nɛ1H (10.12 ppm) resonances (left panel) and upfield-shifted (1.0 ppm–0 ppm) aliphatic proton resonances (right panel).
A gradual sharpening of the proton NMR spectra was observed at pH 4.0, accompanied by reduced chemicalshift dispersions (Fig. 1b ▶), indicating acid-induced denaturation of the partially structured prodomain toward an even more unfolded state. The disappearance of the upfield-shifted methyl resonances at pH 4.0 (Fig. 1b ▶, right panel) strongly indicates the disruption of the (hydrophobic) core structure. The amide proton resonances are restricted to only ∼1 ppm at pH 4.0, which suggests an overall unfolded nature of the prodomain (Fig. 1b ▶, left panel). However, two upfield methyl resonances (at 0.65 ppm and 0.7 ppm) indicate the presence of some residual packing at pH 4.0. The conformational characteristics of the prodomain were further investigated by use of onedimensional proton NMR spectra at additional pH values. At higher pH (pH 7.5–9.0), there were little changes in the broad proton resonances, suggesting the absence of significant conformational changes. Further increase in pH toward more basic conditions caused precipitation of the protein.
The effective hydrodynamic radii (Rh) were measured at pH 6.8 and pH 4.0 using pulse-field-gradient (PFG) NMR at identical concentrations of the prodomains (see Materials and Methods). The values of Rh at pH 6.8 and pH 4.0 were calculated to be 25 Å and 30.5 Å, respectively, indicating again that the broad proton resonances at neutral pH cannot be a result of protein aggregation. More importantly, the increased Rh at pH 4.0 suggests that at pH 6.8 the prodomain has a more compact conformational ensemble than it does at pH 4.0. The compaction factors (see Materials and Methods) were estimated to be 0.52 and 0.14 at pH 6.8 and pH 4.0, respectively. The compaction factor observed here for the prodomain at neutral pH is found to be comparable to those values obtained for the low pH molten-globule states of myoglobin, α-lactalbumin and cytochrome c (Wilkins et al. 1999). It is to be noted that the hydrodynamic radii, or the compaction factors measured for the prodomain, are slightly underestimated because of the presence of flexible tag residues at the termini.
Differential conformational behavior in regions of the prodomain
Figure 2 ▶ compares the 1H-15N HSQC spectra of the prodomain, showing one-bond correlations between the amide proton and the nitrogen-15 resonances. The HSQC spectrum at pH 6.8 (Fig. 2a ▶) is characterized by only a few sharp peaks along with many broad resonances. The extensive broadening for the amide proton resonances (Fig. 1a ▶) at this pH severely reduces the quality of the HSQC spectra with marked broadening and disappearance of many cross peaks, which indicates conformational transitions and averaging that involving most parts of the protein. The sharp resonances in the HSQC spectrum may arise from the flexible tag residues at the N- and C-termini. In contrast, the HSQC spectrum at pH 4.0 has almost all the expected backbone NH correlations (Fig. 2c ▶), making residue-specific assignments possible using standard triple resonance NMR experiments (Bhattacharjya et al. 2000). An interesting feature of the HSQC spectrum at pH 4.0 (Fig. 2c ▶) is that some cross peaks exhibit differential intensities, suggesting that the pH-induced conformational transitions may not be experienced the same way by all the amino acid residues. The relatively broad resonances are from those residues that may undergo slower conformational exchange on the chemical shift scale. On the other hand, the sharper resonances arise from regions of the protein that may be sufficiently unfolded and flexible at pH 4.0. Figure 3 ▶ shows the relative intensities of the HSQC peaks at pH 4.0 of each assigned residue in the prodomain. Strikingly, essentially all the intense peaks are found to be clustered at the C-terminal half of the prodomain that encompasses residues R44– R81. This observation indicates that the entire C-terminal region of the prodomain must have much more flexible conformations than the N-terminal half of the molecule. In contrast, the HSQC peaks from the N-terminal half of the prodomain appear to be more broadened; for example, for residues V3, T7, W8, A9, V10, V18, A19, S21 and A23 (Fig. 2b ▶; Fig.3 ▶), suggesting that the N-terminal part of the prodomain may retain some more ordered structural elements than the C-terminal region. The HSQC peaks cannot even be detected for residues I12, Q33, I34, Y38, Y39, H40 as a result of extensive broadening of these resonances.
Fig. 2.
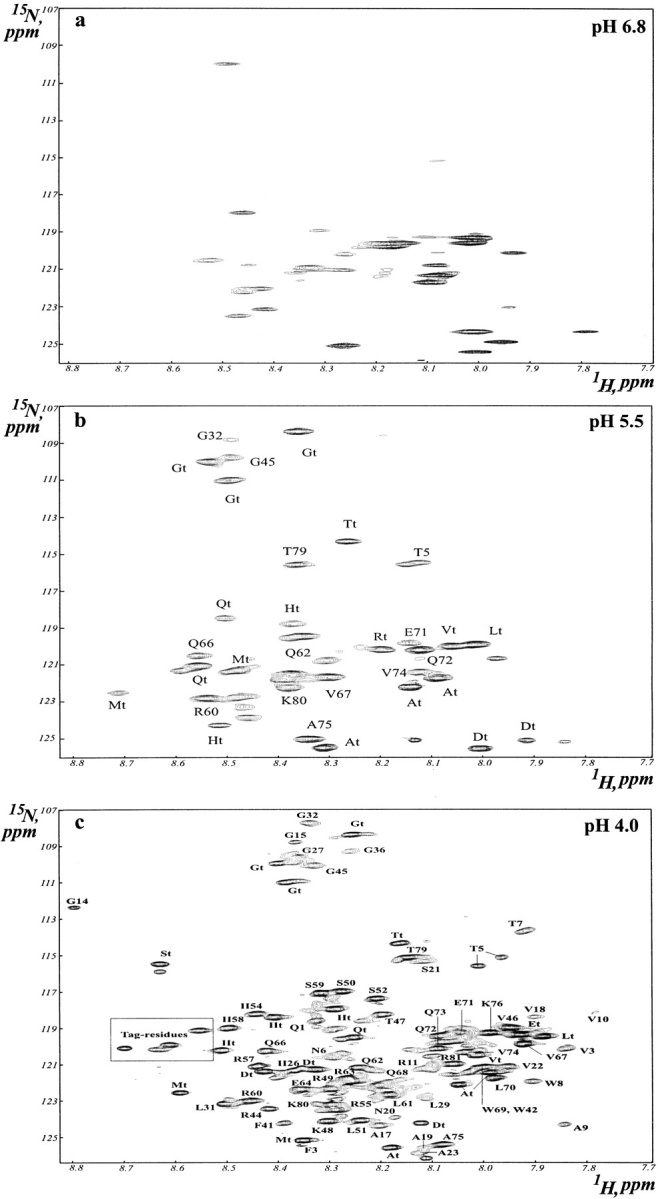
Two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) spectra of the prodomain of furin correlating single bond backbone NH resonances. The HSQC spectra were acquired for the prodomain in 50 mM sodium acetate buffer at 303 K. Some cross peaks that arise from the N- and C-termini tag-residues could be assigned and subscripted with t. The cross peaks inside the box are from the unassigned tag residues.
Fig. 3.

Bar diagram showing intensities of the HSQC cross peaks as a function of residue position of the furin prodomain at pH 4.0. A reliable estimate of intensity could not be obtained for some residues, that is,I12, F28, N30, Q33, I34, F35, Y38, Y39, and H40, from the N terminus either because of signal overlap or extensive broadening of the HSQC peaks.
A 15-residue synthetic peptide (G36-D-Y-Y-H-F-W-H-R-G-V-T-K-R-S50) were synthesized, and its conformational characteristics were studied by use of two-dimensional 1H NMR at both acidic and neutral pH conditions. This peptide fragment did not show any broad resonances at either pHs (spectra not shown) and may therefore adopt an extended or random-coil-like conformation as judged by intense sequential CαH/NH NOEs and the absence of any long- or medium-range NOEs (spectra not shown). In contrast, broad resonances were observed for many residues of this peptide segment at pH 4.0 as part of the full-length prodomain (Fig. 2b,c ▶; Fig. 3 ▶), suggesting that these residues in particular may be involved in long-range interactions along with other residues from the N-terminal region. The residual long-range interactions observed for the N-terminal half of the prodomain at pH 4.0 can be disrupted by the presence of a denaturant, such as 4 M urea as evidenced by uniformly sharp resonances under this condition (spectra not shown).
pH dependence of the conformational flexibility of the prodomain
To gain further insights into the pH-induced conformational transitions of the prodomain, HSQC spectra were recorded at an intermediate pH 5.5 (Fig. 2b ▶) under which more cross peaks can be observed than neutral pH conditions. These HSQC peaks were assigned by a combination of 3-D NOESY-HSQC and TOCSY-HSQC spectra and also by comparison with the HSQC spectra at pH 4.0. Some cross peaks still cannot be assigned to particular residues because of the lack of sequential NOEs. However, most of the cross peaks are found to be from the tag residues that are flexible at all pHs, thereby providing an internal reference. Apart from these easily assigned cross peaks, others can be identified as from residues Q66, V67, E71, Q72, Q73, V74, A75, and T79. Interestingly, none of the residues from the first half of the C-terminal region could be observed in the HSQC spectrum at pH 5.5 except for G45 from the second cleavage site, which indicates increased conformational flexibility around that region. Again, most of the residues from the N-terminal region were not observable apart from T5 and G32 even at a lower pH of 5.5, which indicates that they still have severe line-broadening effects as a result of slow conformational exchanges. These observations taken together show that many of the residues from the first half of the C-terminal region may be involved in slow conformational fluctuations along with the residues from the N-terminal part of the prodomain in a more structured environment.
To delineate the relationship between the N- and C-terminal regions of the prodomain, a 38-residue synthetic peptide that encompasses residues R44–R81 was synthesized, and its conformational characteristics were characterized by use of proton NMR. Figure 4a ▶ shows the aliphatic region of the one-dimensional proton NMR spectra of this peptide at pH 6.8. Appearance of sharp proton resonances indicates that this peptide fragment does not undergo any slow/intermediate conformational exchange. Long-range connectivities that correlate the NH protons to the backbone and side-chain protons can be clearly seen in a TOCSY spectrum (Fig. 4b ▶). The very low chemical shift dispersions of 0.5 ppm both for the amide and the CαH protons also suggest that this peptide fragment is preferentially unfolded at neutral pH. There was no significant change in the NMR spectra at pH 4.0, indicating the absence of any acid-induced conformational transitions for this peptide fragment (spectra not shown).
Fig. 4.
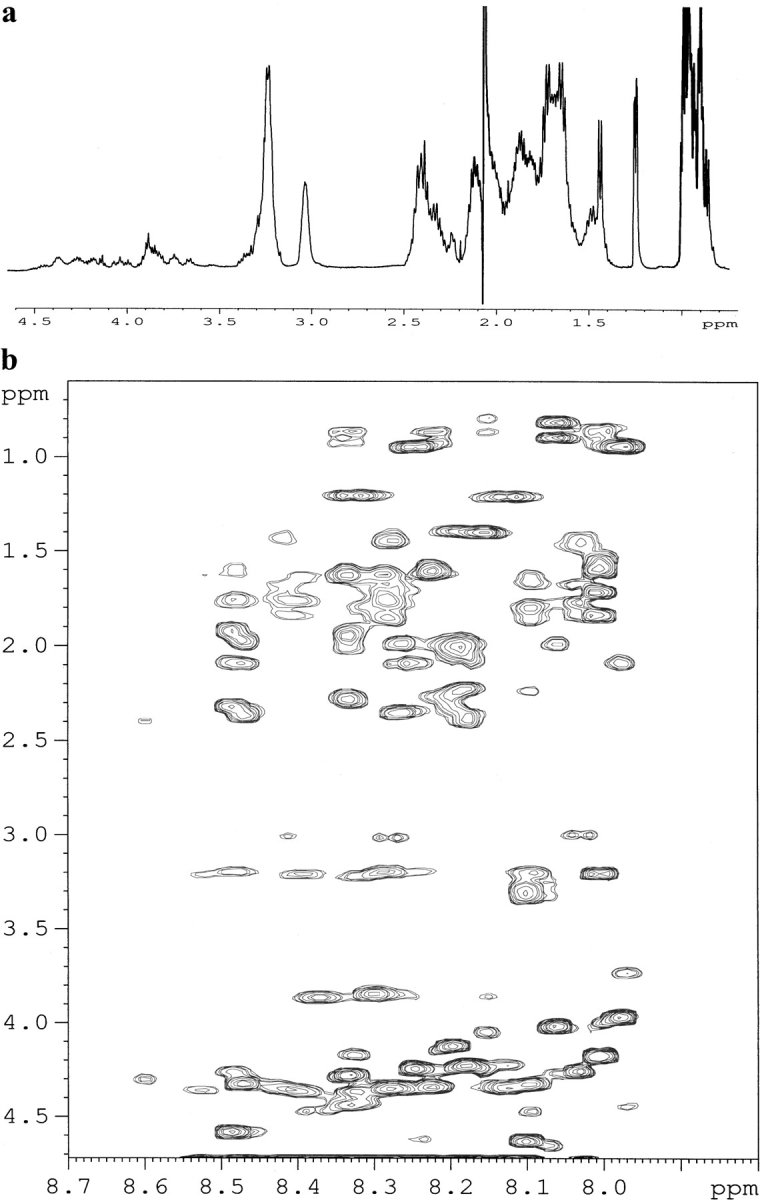
(a) Aliphatic region of the one-dimensional proton NMR spectrum of the C-terminal peptide fragment encompassing residues R44–R81 in 50 mM sodium acetate, pH 6.8, 303 K. (b) Region of the TOCSY spectrum of the same peptide under identical conditions, indicating correlations between the amide proton, CαH proton and other side chain proton resonances.
In summary, these results show that the furin prodomain undergoes a conformational transition from a compact molten-globule like state at neutral pH to a more unfolded state at acidic pH. This pH-induced unfolding transition is predominantly experienced by the residues from the C-terminal region. In contrast, the N-terminal region appears to be relatively more resistant to denaturation. Analysis of HSQC cross peaks at pH 5.5 indicates that the last half of the C-terminal region is more sensitive to pH change as compared with the residues at the beginning. The absence of exchange-induced line broadening in the proton NMR spectra of a peptide fragment from the entire C-terminal region strongly suggests that many residues from the C-terminal region are very likely to be involved in long-range interactions with the N-terminal region of the full-length prodomain.
Discussion
A molten-globule-like state of the furin prodomain under physiological conditions
The full-length prodomain of furin, in isolation and at neutral pH, appears to be `partially folded' and undergoes a slow (microsecond to millisecond time scale) structural fluctuations over different conformational states, causing extensive broadening of the NMR signals. The partially folded state shows significant compactness, with elements of secondary structures and persistent side chain–side chain interactions. Similar conformational characteristics have also been identified in the molten-globule states of many other proteins, for example, in α-lactalbumin (Baum et al. 1989; Alexandrescu et al. 1993; Kim et al. 1999). Detailed characterization using NMR and other methods has established that the molten-globule state of α-lactalbumin is highly compact and possesses secondary structures and a less rigidly packed hydrophobic core (for review, see Ptitsyn 1995). Therefore, the biologically active form of the furin prodomain must also have a partially folded conformation, one that is presumably molten-globule-like. The prodomain of subtilisin, an enzyme belonging to the same family as the convertase, also does not possess a well-folded conformation under biologically active conditions (Eder et al. 1993; Hu et al. 1996). Prodomains from other classes of proteases including cathepsin B (Yu et al. 1998), cathepsin S (Maubach et al. 1997) and carboxypeptidase Y (Sorenson et al. 1993) show analogous conformational characteristics, suggesting that many zymogen prodomains may assume partially folded conformational states.
Recently, it has been shown that many biologically active proteins or protein domains do not have folded three-dimensional structures under physiological conditions, hence they are termed `natively unfolded' (for review, see Wright and Dyson 1999). This class of proteins is mainly involved in protein–protein interactions that control important cellular events, such as cell-cycle regulation, signal transduction, transcription, translation, export across the channels and immunogenecity (Kriwakcki et al. 1996; Daughdrill et al. 1997; Fletcher et al. 1998; Penkett et al. 1998). Conformational analyses of these protein modules by use of NMR have so far shown that they are highly dynamic, noncompact, and devoid of significant side-chain-packing interactions (Kriwakcki et al. 1996; Fletcher et al. 1998; Penkett et al. 1998). In some instances, only transient secondary structure elements can be detected for these proteins (Daughdrill et al. 1997; Hua et al. 1998). In marked contrast with the previous studies, persistent side chain–side chain interactions, secondary structures, and significant compactness were observed in the furin prodomain under physiological conditions, showing that `natively unfolded' proteins can also possess elements of protein-like interactions. In addition, opposed to the notions that the unfolded conformations can recognize diverse biological targets, the furin prodomain shows only limited target selections and exhibits high inhibitory activities only to furin and PC5, an enzyme closely that is related to furin (Zhong et al. 1999). Indeed, selective inhibitions mediated by the prodomains in other enzymatic systems are also well documented (Khan and James 1998).
The restricted target selections by the furin prodomain and other prodomains as well could be dictated by the persistent structural orders of the free states. The ability to recognize a large number of biological targets by the natively unfolded protein domains has been postulated as a result of an `inducible' binding mechanism that must be driven by a large and negative binding enthalpy (Parker et al. 1999; Wright and Dyson 1999). In contrast, target recognitions by well-folded proteins are a `constitutive' one, which is determined by a favorable gain in enthalpy without significant loss in conformational entropy (Wright and Dyson 1999). We propose that the specific and tight binding that is exhibited by the furin prodomain and other prodomains to the cognate proteases may be a result of `semiconstitutive' binding mechanism. In such a situation, the residual structural elements in the partially folded state, most importantly those involving side-chain-packing interactions, could play critical roles in defining the specificity and reducing the entropy costs for binding to the target proteins. Indeed, NMR 15N spin relaxation measurements of the binding of the S-peptide to the S-protein (Alexandrescu et al. 1998) and the folding–unfolding transitions of the SH3 domain (Yang et al. 1997) and staphylococcal nuclease (Yang et al. 1997) have shown that regions with native-like nonlocal interactions in the partially folded states experience the smallest entropy changes on binding or folding. Presence of long-range order in highly dynamic conformational states, such as that assumed by the furin prodomain, can also help retaining excess water molecules inside the "protein core" (Kharakoz and Bychkova 1997; Denisov et al. 1999), which is another significant entropy advantage for target binding that can be realized if these excess water molecules are released (Bogan and Thorn 1998) upon formation of protein–protein complexes.
Acid-induced conformational transitions in the prodomain
Previous in vivo studies have shown that the cleavage of the furin prodomain is an autocatalytic and intramolecular process, whereby the proteolytic cleavage occurs at the consensus cleavage site or the C-terminal end of residues R78-T-K-R81∼ of the prodomain (Leduc et al. 1992; Creemers et al. 1995). These studies have also shown that the cleavage at this potential site is a prerequisite for efficient transport of furin from the endoplasmic reticulum to the trans-golgi network. Recently, a multistep activation pathway of the furin zymogen has been deduced, showing that, after the first cleavage of the prodomain, a second cleavage at the R44-G-V-T-K-R49∼ site in the middle of the prodomain under a more acidic environment is critical for the activation of furin (Anderson et al. 1997). The cleaved prodomain may remain noncovalently associated with furin at neutral pH as shown by coimmunoprecipitation (Anderson et al. 1997) and by the observation that a peptide fragment from the last 10 residues (Q72QVAKRRTKR81) of the prosegment retains some inhibition to the furin enzymatic activity (Zhong et al. 1999). The pH-induced multiple cleavages of the prodomain have also been described for another enzyme PC2 of the same enzyme family (Braks and Martens 1994; Matthews et al. 1994) and may be a common mechanism for the activation of other convertases. Several possible conformational changes can be responsible for this pH-dependent activation process, namely a major structural rearrangement of the protease itself, dissociation of the cleaved prodomain from the enzyme surface caused by destabilization of the interactions between the prodomain and the protease domain, or simply an unfolding of the proregion at acidic pH, enhancing proteolysis.
Here, we show that at neutral pH the isolated prodomain has a compact molten-globule-like conformation with a semirigid protein core. In response to lowering pH, there are major conformational changes occurring within the prodomain. The lowered stability of the core structure at low pH strongly indicates that the involvement of ionic interactions among the residues as the C-terminal part of the prodomain is highly polar, containing many charged amino-acid residues and only a few hydrophobic and aromatic residues. In contrast, there are many aliphatic, aromatic, and a cluster of aromatic residues, Y37-Y-H-F-W-H42, along with some positively charged residues in the N-terminal region of the prodomain. This typical sequence feature for the furin prodomain is shown to be highly conserved in the convertase family (Siezen et al. 1995), indicating that other prodomains may have similar conformational characteristics. The second cleavage site, R44-G-V-T-K-R49∼, close to the aromatic cluster, may be effectively sequestered inside the core structure at neutral pH and thereby not well accessible for proteolytic cleavage. Lowering of pH may result in disruptions of the stabilizing ionic interaction(s) that lead to complete denaturation of the C-terminal half of the molecule and causes the second cleavage site to assume a flexible conformation, thereby promoting a facile cleavage of the prodomain.
Materials and methods
Expression and purification of the furin prodomain
The furin prodomain was overexpressed and purified by use of the Escherichia coli strain BL21 (DE3), using the procedure published elsewhere (Zhong et al. 1999). Apart from the 81 residues of the prodomain, the expression construct also contains an extension of a few residues at both the N and C termini, including a six-residue His-tag for Ni+2 affinity purification, leading to the final amino acid sequence of the expressed protein as MASMTGGQQMGRDP- (Q1KV—KR81)- DVAAALE-(H)6. The expressed prodomain with the additional residues is highly active as a selective inhibitor against the enzymatic activity of furin both in vitro and ex vivo (Zhong et al. 1999). For the production of 15N-labeled or 15N/13C-labeled prodomain, the expressing bacteria were grown in the M9 minimal medium supplemented with either (15NH4)2SO4, or (15NH4)2SO4/13C-glucose. The cells were grown to an OD600 of ∼0.6 and induced by isopropyl β-D-thiogalactoside (IPTG) at a final concentration of 0.5 mM, and grown for another 6 hr before harvesting by centrifugation. Cell pellets were resuspended in a buffer containing 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 5 mM imidazole, followed by sonication and centrifugation. Because the expressed prodomain forms inclusion bodies, cell pellets were redissolved in the above-mentioned buffer containing 8 M urea. The cell lysate thus obtained was loaded onto a Ni-NTA resin and the protein was eluted with high concentrations of imidazole. Fractions containing the prodomain were extensively dialyzed against a 50 mM sodium-acetate buffer at pH 4.0 to remove urea. Finally, samples for NMR analyses were prepared in a solution of 50 mM sodium-acetate, containing 10% D2O and a cocktail of protease inhibitors, phenylmethylsulfonyl fluoride, leuptin, and pepstain. The protein concentrations were estimated to be ∼0.8 mM for samples used for NMR measurements.
Peptide synthesis and purification
Peptides were synthesized using standard FMOC chemistry on an Applied Biosystems 431A synthesizer. The synthetic peptides were purified on an using a preparatory C18 column with a linear gradient of acetonitrile/water mixture containing 0.1% tri-fluoroacetic acid. The major peak fraction was collected and lyophilized. The masses of the peptides were verified by use of electrospray mass spectrometry. Peptide samples for NMR experiments were prepared in 50 mM sodium acetate at pH 6.8. The peptide concentrations were typically 1.5 mM, and the experimental temperatures were fixed at 303 K.
NMR experiments
All the NMR experiments were performed on a Bruker Avance-500 or a Bruker Avance-800 spectrometer equipped with (PFG) units. The water solvent resonance was suppressed by the WATERGATE method (Piottio et al. 1992). A purging field gradient pulse and a water flipback pulse (Fulton and Ni 1997) were employed for the acquisition of the two-dimensional NOESY and TOCSY data in aqueous solution for the peptides. The mixing times were 250 ms for NOESY experiments and 68 ms for TOCSY executed with the TOWNY-16 (Kadkhodaei et al. 1993) mixing sequence. Typically, free induction decays were collected with 2048 data points with 256–512 t1– increments. The NMR data were processed using the XWINNMR software, using π/2 or π/4 cosine window functions along both time dimensions. The detail of all the triple resonance experiments that were performed for the resonance assignments of the prodomain at pH 4.0 have been described elsewhere (Bhattacharjya et al. 2000). The 15N-edited 3-D NOESY-HSQC, and 3-D TOCSY-HSQC experiments at pH 5.5, were performed on the Avance-500 spectrometer with mixing times of 200 ms and 50 ms for the NOESY and TOCSY periods, respectively. For the 3-D experiments, the data sets typically consisted of 96, 30 and 1024 complex points in t1, t2, and t3, respectively. The NMR data were processed using NMRPipe (Delaglio et al. 1995) and analyzed by use of NMRView (Johnson and Blevins 1994).
The hydrodynamic radii of the prodomain were measured at pH 6.8 and pH 4.0 by PFG NMR-diffusion measurements based on the method described by Wilkins et al. (1999) except that CH3COO− was used here as an internal reference molecule. Diffusion experiments were performed with the PG-SLED pulse sequence employing diffusion gradients along the (transverse) x-axis. The lengths of all pulses and delays were held constant, with only the gradient strengths varied from 6% to 100% of their maximum values. The decay of the 1H resonance signals were fitted to a single Gaussian expression: I(g) = Ae-dg2, where I, g, and d represent intensity of the signals, gradient strengths, and decay rates, respectively. The hydrodynamic radii were calculated using the equation Rhprot = dref/dprot (Rhref; Wilkins et al. 1999) where Rhprot is the protein hydrodynamic radius and Rhref is the hydrodynamic radius of the reference molecule, CH3COO−, dref and dprot are the decay rates for the reference molecule and the protein, respectively. The hydrodynamic radius 2.3 Å of CH3COO− was calculated following the relation, Rg = (3/5)1/2 Rh, correlating the radius of gyration (Rg) with Rh (Wilkins et al. 1999). The hydrodynamic radius of the native state of hen lysozyme was estimated here to be 20 Å using 2.3 Å as Rhref, which is in excellent agreement with the previous estimation of Rh for lysozyme using dioxan as a reference molecule (Wilkins et al. 1999). The compaction factors (C) for the prodomain were obtained from the hydrodynamic radii following the relationship, C = RhD−Rh / RhD− RhN where RhD and RhN are the predictive values for the hydrodynamic radii of the native and fully denatured states, respectively, and Rh is the experimentally measured hydrodynamic radius (Wilkins et al. 1999).
Acknowledgments
This work was supported in part by the Protein Engineering Network Centres of Excellence, sponsored by the Government of Canada, by a Medical Research Council Canada Program Grant (No. PG-11474 to N.G.S. and M.C.), and by the National Research Council of Canada (NRCC Publication No. 42998).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
PCs, proprotein convertases
NMR, nuclear magnetic resonance
NOESY, nuclear Overhauser effect spectroscopy
TOCSY, total correlation spectroscopy
HSQC, heteronuclear single quantum coherence
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.41301.
References
- Alexandrescu, A.T., Evans, P.A., Pitkeathly, M., Baum, J., and Dobson, C.M. 1993. Structure and dynamics of the acid-denatured molten globule state of alpha-lactalbumin: A two-dimensional NMR study. Biochemistry 32 1707–1718. [DOI] [PubMed] [Google Scholar]
- Alexandrescu, A.T., Rathgeb-Szabo, K., Rumpel, K., Jahnke, W., Schulthess, T., and Kammerer, R.A. 1998. 15N backbone dynamics of the S-peptide from ribonuclease A in its free and S-protein bound forms: Toward a site-specific analysis of entropy changes upon folding. Protein Sci. 7 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, E.D., VanSlyke, J.K., Thulin, C.D., Jean, F., and Thomas, G. 1997. Activation of the furin endoprotease is a multiple-step process: Requirements for acidification and internal propeptide cleavage. EMBO J. 16 1508–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, J., Dobson, C.M., Evans, P.A., and Hanley, C. 1989. Characterization of a partly folded protein by NMR methods: studies on the molten globule state of guinea pig α-lactalbumin. Biochemistry 28 7–13. [DOI] [PubMed] [Google Scholar]
- Bhattacharjya, S. and Balaram, P. 1997. Effects of organic solvents on protein structures: observation of a structured helical core in hen egg-white lysozyme in aqueous dimethylsulfoxide. Proteins: Struct. Funct. and Genet. 29 492–507. [DOI] [PubMed] [Google Scholar]
- Bhattacharjya, S., Ping, X., and Ni, F. 2000. Sequence-specific 1H, 15N and 13C resonance assignments of the inhibitory prodomain of human furin. J. Biomol. NMR 16 275–276. [DOI] [PubMed] [Google Scholar]
- Braks, J.A. and Martens, G.J. 1994. 7B2 is a neuroendocrine chaperone that transiently interacts with prohormone convertase PC2 in the secretory pathway. Cell 78 263–273. [DOI] [PubMed] [Google Scholar]
- Bogan, A.A. and Thorn, K.S. 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280 1–9. [DOI] [PubMed] [Google Scholar]
- Chapman, R.L., Kane, S.E., and Erickson, A.H. 1997. Abnormal glycosylation of procathepsin L due to N-terminal point mutations correlates with failure to sort to lysosomes. J. Biol. Chem. 272 8808–8816. [DOI] [PubMed] [Google Scholar]
- Creemers, J.W., Vey, M., Schafer, W., Ayoubi, T.A., Roebroek, A.J., Klenk, H.D., Garten, W., and Van de Ven, W. J. 1995. Abnormal glycosylation of procathepsin L due to N-terminal point mutations correlates with failure to sort to lysosomes. J. Biol. Chem. 270 2695–2702.7852339 [Google Scholar]
- Daughdrill, G.W., Chadsey, M.S., Karlinsey, J.E., Hughes, K.T., and Dahlquist, F.W. 1997. The C-terminal half of the anti-σ factor, FlgM, becomes structured when bound to its target sigma 28. Nat. Struct. Biol. 4 285–291. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6 277–293. [DOI] [PubMed] [Google Scholar]
- Denisov, V.P., Jonsson, B.H., and Halle, B. 1999. Hydration of denatured and molten globule proteins. Nat. Struct. Biol. 6 253–260. [DOI] [PubMed] [Google Scholar]
- Eder, J., Rheinnecker, M., and Fersht, A.R. 1993. Folding of subtilisin BPN': Role of the pro-sequence. J. Mol. Biol.. 233 293–304. [DOI] [PubMed] [Google Scholar]
- Eliezer, D., Yao, J., Dyson, H.J., and Wright, P.E. 1998. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat. Struct. Biol. 5 148–155. [DOI] [PubMed] [Google Scholar]
- Erickson, A.H. 1989. Biosynthesis of lysosomal endopeptidases. J. Cell Biochem. 40 31–41. [DOI] [PubMed] [Google Scholar]
- Fletcher, C.M., McGuire, A.M., Gingras, A.C., Li, H.J., Matsuo, H., Sonenberg, N., and Wanger, G. 1998. 4E binding proteins inhibit the translation factor eIF4E without folded structure. Biochemistry 37 9–15. [DOI] [PubMed] [Google Scholar]
- Fuller, R.S., Brake, A.J., and Thorner, J. 1989. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science 246 482–486. [DOI] [PubMed] [Google Scholar]
- Fulton, D.B. and Ni F. 1997. ROESY with water flip back for high-field NMR of biomolecules. J. Magn. Reson. 129 93–97. [DOI] [PubMed] [Google Scholar]
- Hu, Z., Haghjoo, K., and Jordan, F. 1996. Further evidence for the structure of the subtilisin propeptide and for its interactions with mature subtilisin. J. Biol. Chem. 271 3375–3384. [DOI] [PubMed] [Google Scholar]
- Hua, Q., Jia, W., Bullock, B.P., Habener, J.F., and Weiss, M.A. 1998. Transcriptional activator-coactivator recognition: Nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry 37 5858–5866. [DOI] [PubMed] [Google Scholar]
- Jan, G., Taylor, N.A., Scougall, K.T., Docherty, K., and Shennan, K.I.J. 1998. The propeptide of prohormone convertase PC2 acts as a transferable aggregation and membrane-association signal. Eur. J. Biochem. 257 41–46. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 4 603–614. [DOI] [PubMed] [Google Scholar]
- Kadkhodaei, M., Hwang, T.L., Tang, J., and Shaka, A.J. 1993. A simple windowless mixing sequence to suppress cross relaxation in TOCSY experiments. J. Magn. Reson. A105 104–107. [Google Scholar]
- Khan, A.R. and James, M.N. 1998. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7 815–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharakoz, D.P. and Bychkova, V. E. 1997. Molten globule of human α-lactalbumin: hydration, density, and compressibility of the interior. Biochemistry 36 1882–1890. [DOI] [PubMed] [Google Scholar]
- Kim, S., Bracken, C., and Baum, J. 1999. Characterization of millisecond time-scale dynamics in the molten globule state of α-lactalbumin by NMR. J. Mol. Biol. 294 551–560. [DOI] [PubMed] [Google Scholar]
- Kriwacki, R.W., Hengst, L., Tennant, L., Reed, S.I., and Wright, P.E. 1996. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. 93 11504–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc, R., Molloy, S., Thorne, B.A., and Thomas, G. 1992. Activation of human furin precursor processing endoprotease occurs by an intramolecular autoproteolytic cleavage. J. Biol. Chem. 267 14304–14308. [PubMed] [Google Scholar]
- Mach, L., Mort, J.S., and Glossl J. 1994. Maturation of human procathepsin B. Proenzyme activation and proteolytic processing of the precursor to the mature proteinase, in vitro, are primarily unimolecular processes. J. Biol. Chem. 269 13030–13035. [PubMed] [Google Scholar]
- Matthews, G., Shennan, K.I., Seal, A.J., Taylor, N.A., Colman, A., and Docherty, K. 1994. Autocatalytic maturation of the prohormone convertase PC2. J. Biol. Chem. 269 588–592. [PubMed] [Google Scholar]
- Maubach, G., Schilling, K., Rommerskrich, W., Wenz, I., Schultz, J.E., Weber, E., and Weideranders, B. 1997. The inhibition of cathepsin S by its propeptide-specificity and mechanism of action. Eur. J. Biochem. 250 745–750. [DOI] [PubMed] [Google Scholar]
- McIntyre, G.F. and Erickson, A.H. 1993. The lysosomal proenzyme receptor that binds procathepsin L to microsomal membranes at pH 5 is a 43-kDa integral membrane protein. Proc. Natl. Acad. Sci. 90 10588–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, K. 1997. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 327 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, D., Rivera, M., Zor, T., Henrion-Caude, A., Radhakrishnan, I., Kumar, A., Shapiro, L.H., Wright P.E., Montminy, M., and Brindle, P.K. 1999. Role of secondary structure in discrimination between constitutive and inducible activators. Mol. Cell. Biol. 19 5601–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkett, C.J., Redfield, C., Jones, J.A., Dodd, I., Hubbard, J., Smith, R.A.G., Smith, L.J., and Dobson, C.M. 1998. Structural and dynamical characterization of a biologically active unfolded fibronectin-binding protein from Staphylococcus aureus. Biochemistry 37 17054–17067. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J.. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47 83–229. [DOI] [PubMed] [Google Scholar]
- Roebroek, A.J.M., Schalken, J.A., Bussemakers, M.J.G., Van Heerikhuizen, H., Onnekin, C., Debruyne, F.M.J., Bloemers, H.P.J., and Van de Ven, W.J.M. 1986. Characterization of human c-fes/fps reveals a new transcription unit (fur) in the immediately upstream region of the proto-oncogene. Mol. Biol. Rep. 11 117–125. [DOI] [PubMed] [Google Scholar]
- Ruvinov, S., Wang, L., Ruan, B., Almog, O., Gilliland, G.L., Eisenstein, E., and Bryan P.N. 1997. Engineering the independent folding of the subtilisin BPN' prodomain: Analysis of two-state folding versus protein stability. Biochemistry 36 10414–10421. [DOI] [PubMed] [Google Scholar]
- Seidah, N.G. and Chrétien, M. 1999. Proprotein and prohormone convertases: A family of subtilases generating diverse bioactive polypeptides. Brain Res. 848 45–62. [DOI] [PubMed] [Google Scholar]
- Seidah, N.G. and Chrétien, M. 1997. Eukaryotic protein processing: endoproteolysis of precursor proteins. Curr. Opin. in Biotech. 8 602–607. [DOI] [PubMed] [Google Scholar]
- Seidah, N.G. 1995. In Intramolecular chaperones and protein folding (eds. U. Shinde and M. Inouye), pp. 181–203, R. G. Landes Co., Austin, TX.
- Shinde, U. and Inouyne, M. 1993. Intramolecular chaperones and protein folding. Trends Biochem. Sci. 18 442–446. [DOI] [PubMed] [Google Scholar]
- Siezen, R.J. and Leunissen, J.A. 1997. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 36 6968–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siezen, R.J., Leunissen, J.A.M., and Shinde U. 1995. Homology analysis of the propeptides of subtilisin-like serine proteases (subtilases). In Intramolecular chaperones and protein folding (eds. U. Shinde and M. Inouye), pp. 233–256, R. G. Landes Co., Austin, TX.
- Silen, J.L. and Agard, D.A. 1989. The α-lytic protease pro-region does not require a physical linkage to activate the protease domain in vivo. Nature 341 462–464. [DOI] [PubMed] [Google Scholar]
- Sorenson, P., Winthe, J.R., Kaarsholm, N.C., and Poulsen, F.M. 1993. The pro region required for folding of carboxypeptidase Y is a partially folded domain with little regular structural core. Biochemistry 32 12160–12166. [DOI] [PubMed] [Google Scholar]
- Takeda-Ezaki, M. and Yamamoto, K. 1993. Isolation and biochemical characterization of procathepsin E from human erythrocyte membranes. Arch. Biochem. Biophys. 304 352–358. [DOI] [PubMed] [Google Scholar]
- Takeshima, H., Sakaguchi, M., Mihara, K., Murakami, K., and Omura, T. 1995. Intracellular targeting of lysosomal cathepsin D in COS cells. J. Biochem. (Tokyo) 118 981–988. [DOI] [PubMed] [Google Scholar]
- Valls, L.A., Winther, J.R., and Stevens, T.H. 1990. Yeast carboxypeptidase Y vacuolar targeting signal is defined by four propeptide amino acids. J. Cell Biol. 111 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Ven, W.J., Roebroek, A.J., and Van Duijnhoven, H.L. 1993. Structure and function of eukaryotic proprotein processing enzymes of the subtilisin family of serine proteases. Crit. Rev. Oncog. 4 115–136. [PubMed] [Google Scholar]
- Wilkins, D.K., Grimshaw, S.B., Receveur, V., Dobson, C.M., Jones, J.A., and Smith, L.J. 1999. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 38 16424–16431. [DOI] [PubMed] [Google Scholar]
- Wright, P.E. and Dyson, H.J. 1999. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293 321–331. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of proteins and nucleic acids, John Wiley, New York.
- Yang, D., Mok, Y.K., Forman-Kay, J.D., Farrow, N.A., and Kay, L.E. 1997. Contributions to protein entropy and heat capacity from bond vector motions measured by NMR spin relaxation. J. Mol. Biol. 272 790–804. [DOI] [PubMed] [Google Scholar]
- Yu, Y., Vranken, W., Goudreau, N., de Miguel, E., Mangy, M.C., Mort, J.S., Dupras, R., Storer, A.C., and Ni, F. 1998. An NMR-based identification of peptide fragments mimicking the interactions of the cathepsin B propeptide. FEBS Lett. 429 9–16. [DOI] [PubMed] [Google Scholar]
- Zhong, M., Munzer, J.S., Basak, A., Benjannet, S., Mowla, S.J., Decroly, E., Chrétien, M., and Seidah, N.G. 1999. The prosegments of furin and PC7 as potent inhibitors of proprotein convertases. In vitro and ex vivo assessment of their efficacy and selectivity. J. Biol. Chem. 274 33913–33920. [DOI] [PubMed] [Google Scholar]