

Abstract
The family of three-dimensional molecular structures of the major coat protein from the M13 bacteriophage, which was determined in detergent micelles by NMR methods, has been analyzed by constrained geometry optimization in a phospholipid environment. A single-layer solvation shell of dioleoyl phosphatidylcholine lipids was built around the protein, after replacing single residues by cysteines with a covalently attached maleimide spin label. Both the residues substituted and the phospholipid were chosen for comparison with site-directed spin labeling EPR measurements of distance and local mobility made previously on membranous assemblies of the M13 coat protein purified from viable mutants. The main criteria for identifying promising candidate structures, out of the 300 single-residue mutant models generated for the membranous state, were 1) lack of steric conflicts with the phospholipid bilayer, 2) good match of the positions of spin-labeled residues along the membrane normal with EPR measurements, and 3) a good match between the sequence profiles of local rotational freedom and a structural restriction parameter for the spin-labeled residues obtained from the model. A single subclass of structure has been identified that best satisfies these criteria simultaneously. The model presented here is useful for the interpretation of future experimental data on membranous M13 coat protein systems. It is also a good starting point for full-scale molecular dynamics simulations and for the design of further site-specific spectroscopic experiments.
Keywords: Viral coat protein, M13 bacteriophage, site-directed spin-labeling, molecular modeling, electron paramagnetic resonance, membrane protein, lipid-protein interaction
M13 is a small filamentous bacteriophage that is specific to Escherichia coli. It consists of ∼2800 copies of the coat protein surrounding the genetic material, a circular single-stranded DNA (Marvin and Hohn 1969; Rasched and Oberer 1986). Most of the virus coat (98%) is made up of the gene 8 product that forms a 1.5–2.0 nm thick, flexible cylinder around the viral DNA: the major coat protein. The newly synthesized coat proteins are inserted in the cytoplasmic membrane of the host, where they are stored before being used in the assembly process. The major coat protein is a small multifunctional structural protein, composed of 50 amino acids (Van Wezenbeek et al. 1980). The primary sequence of the major coat protein is specifically tuned for diverse structural roles because it is capable of protein–DNA, protein–protein, and protein–lipid interactions (Hemminga et al. 1993). To accomplish this, the major coat protein has three domains: a positively charged C-terminal domain, which primarily is responsible for the interaction with DNA; a hydrophobic central part of the protein, which stabilizes protein–protein interactions in the virus particle, as well as maintains a stable thermodynamic association with the host membrane during replication; and an amphipathic N-terminal domain, which is thought to be important in docking the protein at the virus assembly site (Marvin et al. 1994).
Knowledge of the three-dimensional structure of the assembly proteins in different stages of the M13 phage life cycle would be essential to understand the molecular details of viral action. Of particular interest is how the phage injects its genetic material into the host cell and then how the newly assembled phage leaves the cell without causing lysis of the host. Determination of the structure of the membranous form of the M13 major coat protein is therefore important. The classical structural methods, namely X-ray and NMR techniques, are subject to well-known limitations when applied to proteins in the membrane-bound state. In the absence of suitable crystals, possibilities to proceed are 1) to solubilize the membrane protein in detergent micelles for NMR studies, or 2) to use less direct spectroscopic structural techniques on the protein in its native membranous form.
A family of structural alternatives has been determined for the M13 major coat protein in both DodPC and SDS detergents (25 in each detergent), based on NMR studies (Papavoine et al. 1998). Although there are experimental indications that the structure of the protein may differ somewhat between detergent micelles and phospholipid bilayers (Stopar et al. 1996), these structures serve as a suitable starting point for modeling the M13 major coat protein in a phospholipid bilayer membrane. In this paper, we have tested these micellar structures against experimental constraints obtained from the protein embedded in a phospholipid bilayer to identify those most compatible with a lipid membrane environment. To achieve this, the structures of the M13 major coat protein were modified with single-residue replacements by a spin-labeled cysteine, and a first lipid-bilayer shell of DOPC was constructed around the protein. These modifications were followed by constrained molecular mechanics geometry optimization, whereby the protein backbone was left unchanged. The experimental constraints were distance data on the membrane topology of spin-labeled mutants (A25C, V31C, T36C, G38C, T46C, and A49C) of the coat protein reconstituted in a phospholipid bilayer, as well as EPR parameters characterizing the rotational freedom (namely outer hyperfine splitting) of the spin label attached at these residues (Stopar et al. 1997). It is found that the sequence profiles of these parameters are sufficiently selective to discriminate between various conformations of the membrane-bound protein. A unique subclass of structure is identified that best fits the available experimental data from membranes.
Results and Discussion
Starting structures
The backbone atoms (N, C, and Cα) of the M13 major coat protein structures in SDS micelles, determined by NMR and optimization techniques (Papavoine et al. 1998), are shown schematically in Figure 1 ▶. These structures were subject to further experimental constraints in the membrane-bound state, which were obtained from viable cysteine mutants in spin-label EPR experiments (Stopar et al. 1997). To do this, we built a spin-labeled cysteine, made single-residue replacements in the M13 major coat protein structures with cysteine-maleimide, optimized the modified structures, and then derived structural parameters from the models for comparison with the experimental EPR data. Some of the structures in Figure 1 ▶ that are possible in small, highly curved micelles can be eliminated immediately as suitable candidates for the structure in planar membranes, because the charged N-terminal section would bend back into the hydrophobic interior of the membrane. These are the U-shaped structures that are indicated by light lines in Figure 1 ▶ and constitute not more than 7–8 members in each set (see Table 1). In addition, solid-state NMR studies on aligned phospholipid bilayers have indicated that the N-terminal helix (specifically the section containing Leu 14) of the closely related fd bacteriophage coat protein is oriented nearly perpendicular to the transmembrane helix (McDonnell et al. 1993). This further justifies elimination of the U-shaped structures.
Fig. 1.

Backbone atoms (N, C, and Cα) for the 25 low-energy structures (with the backbone aligned between residues 25 and 45) of the M13 major coat protein in SDS micelles. Structures were obtained from the Brookhaven protein data bank (entry code 2CPS) as published by Papavoine et al. (1998). Thin lines indicate U-shaped configurations that are incompatible with a planar membrane environment. The complete amino acid sequence is AEGDDPAKAAFNSLQASATEYIGYAWAMVVVIV GATIGIKLFKKFTSKAS.
Table 1.
Angle between transmembrane and N-terminal helices in NMR-derived structures of M13 major coat protein in DodPC (PDB: 2CPB) and SDS (PDB: 2CPS) micelles determined by Papavoine et al. (1998)a
| Structure no. | DodPC | SDS |
| 1 | 146° | 100° |
| 2 | 117° | 129° |
| 3 | 99° | 139° |
| 4 | 69° | 30° |
| 5 | 123° | 111° |
| 6 | 70° | 90° |
| 7 | 117° | 122° |
| 8 | 59° | 17° |
| 9 | 64° | 63°b |
| 10 | 80° | 69°b |
| 11 | 35° | 108° |
| 12 | 52° | 146° |
| 13 | 150° | 113° |
| 14 | 121° | 111° |
| 15 | 132° | 61°b |
| 16 | 153° | 69° |
| 17 | 100° | 85° |
| 18 | 50° | 147° |
| 19 | 18° | 154° |
| 20 | 96° | 81° |
| 21 | 65°b | 56° |
| 22 | 59° | 51° |
| 23 | 150° | 93° |
| 24 | 92° | 125° |
| 25 | 44° | 91° |
a Structures with an angle between helix axes of <60° were discarded in the model building.
b These structures were also discarded because the non-helical part of the N-terminal forms a U-shape.
Spin label structure
The 5-MSL spin label was built and minimized as the two stereoisomers arising from the chiral carbon in the proxyl ring. After connecting 5-MSL to the sulphydryl group of the cysteine residue, four stereoisomers were obtained. The eight structures obtained by rotating the proxyl ring around the N–C bond by 180° for each stereoisomer were optimized using MMFF94. One structure was chosen (Fig. 2 ▶) on the basis of lowest energy. Only two other of the alternative structures are expected to be comparably populated at room temperature. The remainder were predicted to lie higher in energy by 1.3–3.6 kcal/mol. In the chosen structure, the long axis of the maleimide spin label is oriented approximately perpendicular to the helix axis, in agreement with previous suggestions (Wolkers et al. 1997).
Fig. 2.

Optimized structure of spin-labeled cysteine. The 5-maleimidoproxyl spin label that is covalently attached at the sulphydryl group (S) is indicated together with the nitroxide group (N–O) of the proxyl ring. The structure was built and optimized in Spartan with the MMFF94 force field, and is displayed using Insight II.
Spin-labeled protein without lipid
The spin-labeled cysteine (Fig. 2 ▶) was substituted in the original NMR structures of Figure 1 ▶, and in the corresponding ones from DodPC, at positions corresponding to the single cysteine mutations (A25C, V31C, T36C, G38C, T46C, and A49C) used in the spin-label EPR studies (Stopar et al. 1997). The residue replacements were performed in MOLMOL and are indicated, on a single peptide backbone, in Figure 3 ▶. Excluding U-shaped conformations, this generates a total of ∼35 possible structures for each mutant. For mutation positions definitely within the hydrophobic region (residues 31, 36, and 38), the number of independent structures was reduced to minimally 26 by grouping together equivalent structures. These are representative, on the basis of rms differences, of the local structures ±2 residues from the mutated residue. (It was verified that the structures assigned as equivalent are not differentially influenced by the presence of solvating phospholipids.) Because structures at the ends of the hydrophobic region are not well aligned within a family (see Fig. 1 ▶), all 35 possibilities were treated explicitly for mutated residues 25, 46, and 49, which are expected to be located close to the lipid headgroup regions. The protein-attached spin label structures were then reoptimized using the MMFF94 force field.
Fig. 3.

Ribbon representation of the M13 major coat protein structure no. 20 of entry 2CPS from the Brookhaven Protein Data Bank (Papavoine et al. 1998). Side chains of the original residues that were changed singly to cysteine (top to bottom: A25C, V31C, T36C, G38C, T46C, and A49C), both by mutagenesis (Stopar et al. 1997) and by modeling in the present study, are shown in ball-and-stick representation. The figure was created using MOLMOL (Koradi et al. 1996).
The resulting trial structures could then be compared with the EPR results on the different spin-labeled mutant proteins reconstituted in phospholipid membranes. The relevant experimental data are not only measurements of the vertical position of the spin-labeled residue in the membrane, but importantly are also the profile of local mobility of the spin label with position in the sequence (Stopar et al. 1997). The latter is parameterized by the outer hyperfine splitting, 2Amax, in the spin-label EPR spectrum (see, e.g., Marsh 1981). The experimental profile is characterized by a remarkably high value of 2Amax at Cys 25, which decreases nonmonotonically on proceeding further along the sequence.
The local rotational mobility of the spin label will depend on the atomic packing density in the region of attachment of the spin label. We have chosen to characterize this packing density by a parameter (f) that is defined by
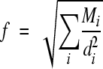 |
1 |
where mi is the mass and di is the distance (in Å) of the ith atom measured from the reference atom. The reference position was the nitrogen in the proxyl ring of 5-MSL. All atoms of the protein, including hydrogens, were used in the summation. Only the atoms of the spin label were excluded from calculation of the f-parameter. The f-parameter in Equation 1 differs slightly from a previous one that was used successfully in a similar site-directed spin-labeling study on cytochrome c in solution (Turyna et al. 1998). (Note that the latter reference contains a printing error.) The extensions made here are to include mass weighting and to retain hydrogen atoms in the summation. Note that it is only relative values of Amax (i.e., the shape of the profile) with which we seek to correlate the f-parameter. For this, a qualitative correlation should suffice. However, the final fit (see Fig. 8 ▶, below) does imply an approximately linear relation, most probably because the spectra analyzed are confined to the same motional regime of spin-label dynamics (see, e.g., Marsh and Horváth 1989).
Fig. 8.

Sequence profiles of the f-parameter from cysteine–maleimide mutants of the M13 major coat protein at different vertical shifts (broken lines), for the structure shown in Fig. 6 ▶ that was optimized with zero shift. The heavy solid line gives the sequence profile for the final structure that is reoptimized with +1 Å vertical shift. Circles are the experimental outer hyperfine splitting profile that is scaled linearly (with offset) for best match to the f-parameter profile of the final (+1 Å-shifted) structure.
The dependence of the f-parameter on the range over which the summation is made is shown in Figure 4 ▶. Data are given for three different mutants, and the effect of including or excluding hydrogen atoms is also shown. A 9-Å range was finally chosen for the summation, after testing various summations ranging from 4 to 50 Å, because this was found to give near-optimal discrimination between the different mutants (see Fig. 4 ▶). The 9-Å radial region is large enough to include all atoms that can affect the spin label directly and is small enough to exclude atoms that have no direct influence on the spin label.
Fig. 4.

Dependence of the packing parameter (f, defined in Eq. 1) on the radial region over which the atom summation is made. Results of calculations are given for spin-labeled cysteine mutants: A25C (circles), V31C (squares), and A49C (triangles), either with (solid symbols) or without (open symbols) hydrogens included in the evaluation. The radius of the region was measured from the reference atom, which was the nitrogen atom of the proxyl ring of the spin label.
The profiles of the calculated f-parameters are shown in Figure 5 ▶ for the two reduced sets of structures (i.e., from the families of structures in DodPC and SDS). Relative to the experimental EPR profile, the most significant feature of Figure 5 ▶ is that only structures in which the Cys 25 residue is inside the interhelix hinge region between the N-terminal and transmembrane segments (cf. Fig. 3 ▶) give rise to a significantly higher f-parameter than do the other mutant positions. This substantiates previous suggestions from EPR and molecular modeling studies on the hinge region (Wolkers et al. 1997). The structures used in Figure 5 ▶ are still without the lipid shell, but it was found subsequently that the large f-parameter of Cys 25, relative to other positions, could not be produced simply by adding lipids (data not shown). Therefore, this finding limits suitable candidates to those structures that are L-shaped (see Table 1) and for which Cys 25 is inside the hinge region. Surprisingly, no such structure was found in the DodPC family (i.e., PDB: 2CPB; Fig. 5 ▶, top). Out of the small subset (3–4) of very similar structures found to fulfill this condition in the SDS family (i.e., PDB: 2CPS), structure no. 20 was selected as representative for further model building because the f-parameter profile (see Fig. 5 ▶, bottom) most resembled the experimental mobility profile. In this subset of structures, the large f-parameter (and outer hyperfine splitting) for residue 25 is caused by the cysteine–maleimide being squeezed between Trp 26 and Ile 22 side chains. Additionally, putative hydrogen bonds between Trp 26 and maleimide oxygens possibly contribute to immobilization of the spin label. Structure no. 20 from the SDS family was a favorable choice also because the C-terminal half of the hinge region (residues 17–24) of the protein is better defined in SDS micelles than in DodPC micelles (Papavoine et al. 1998).
Fig. 5.
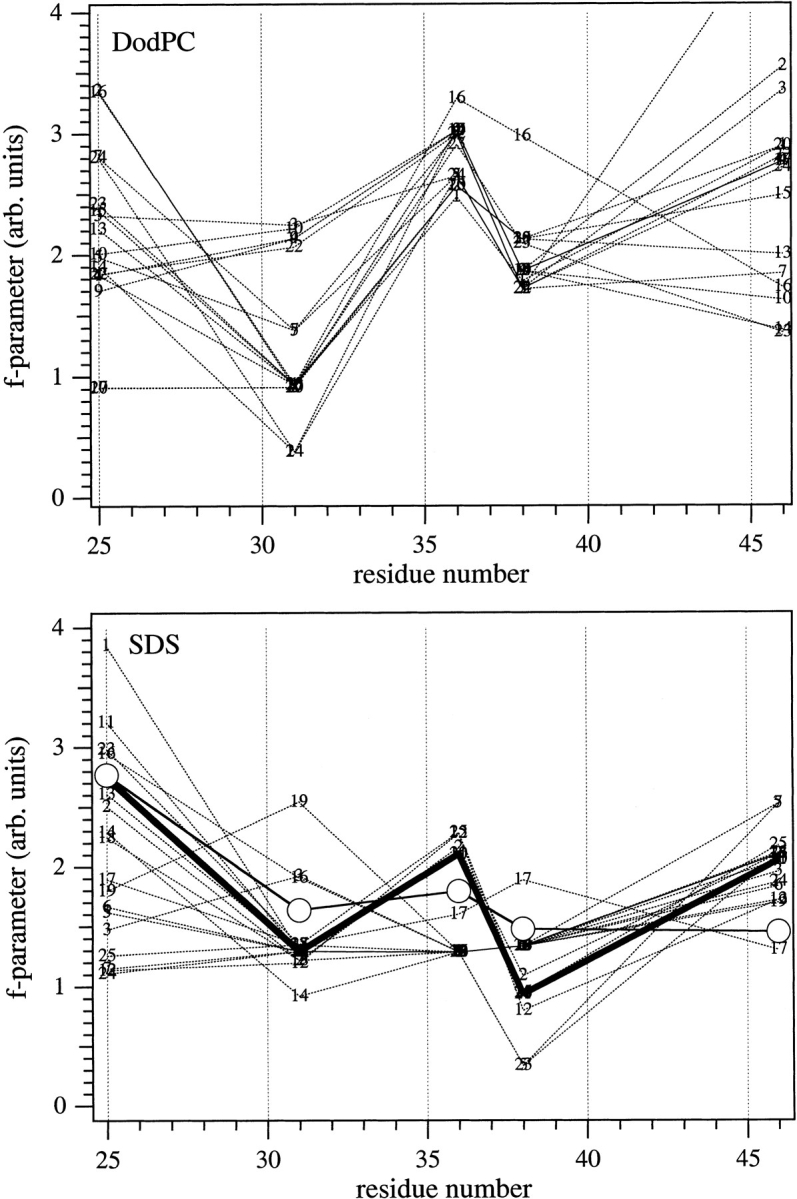
Sequence profiles for the f-parameter of M13 coat protein structures with single cysteine–maleimide replacements, in the absence of a lipid bilayer. Each x-coordinate represents a single mutant of the original families of structures determined in DodPC (top) or in SDS (bottom), in which the cysteine–maleimide side chain was locally optimized after amino acid replacement. Structure numbers indicated in the figure are those in the PDB files 2CPB and 2CPS for NMR-based structures in DodPC and SDS micelles, respectively, determined by Papavoine et al. (1998). (Bottom) The heavy solid line indicates the selected structure no. 20; open circles are the experimental EPR outer hyperfine splittings scaled to best match this profile.
Spin-labeled protein with lipids
A single shell (the first solvation bilayer shell) of DOPC lipids was constructed around the selected M13 major coat protein structure. First, a single DOPC molecule was built within Spartan. The glycerol region was created according to the sc/γ structure given by Pascher (1996). The electrostatic charges for 5-MSL and DOPC were calculated using density functional theory at the pBP86/DN** level. The predominantly all-trans fatty acid chains of the DOPC molecule were shortened using the tether tool in Sculpt to match the apolar region of the protein. A lipid shell was then constructed in MOLMOL using this DOPC structure, which resulted in a bilayer thickness of ∼33 Å between phosphorus atoms of opposing molecules. This is close to the length of the intramembranous part of the protein, which is 31.8 Å between the α carbons of residues 25 and 46 of structure no. 20 from PDB entry 2CPS. For comparison, the phosphate–phosphate distance in a fully hydrated DOPC bilayer is ≈35.3 Å, and the hydrocarbon thickness is 27.2 Å (Tristram-Nagle et al. 1998).
After a test minimization of the M13 protein surrounded by 18 DOPC molecules in Sculpt, the number of lipid molecules was reduced to 12 (one shell of six lipids for each bilayer half). The diameter of this lipid shell was ∼19 Å. Each of the 6 mutants of structure no. 20 was inserted in the center of the lipid shell in MOLMOL, with vertical positioning of the hydrophobic helix according to the distance measurements of Stopar et al. (1997). This composite structure was minimized in Sculpt with only the protein backbone frozen. Additional forces (called springs in Sculpt) were applied to every atom of the lipid molecules, in a direction towards the axis of the protein hydrophobic helix. The MM3 force field was used and Van der Waals interactions were modeled with a modified Lennard-Jones potential between atoms within 6 Å of each other (Surles et al. 1994). Electrostatic interactions were treated with a Coulomb model, using a distance-dependent dielectric constant between atoms within 10 Å of each other (Surles et al. 1994). These structures were optimized until the fractional change in energy was <0.01. The resulting optimized model is shown for the spin-labeled A25C mutant in Figure 6 ▶.
Fig. 6.
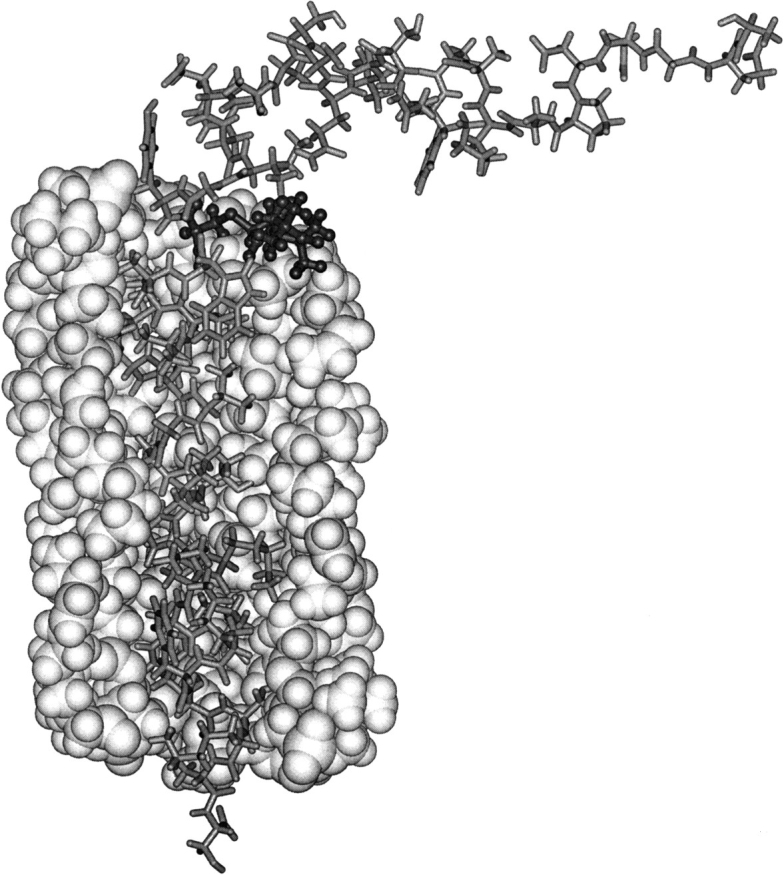
Structure of Cys 25-spin labeled M13 coat protein (stick representation) surrounded by a single bilayer solvation shell of DOPC phospholipids. For clarity, only part of the lipid shell is shown—in space-filling representation. The structure was chosen to satisfy the experimental EPR constraints and was obtained by geometry optimization of the lipids and all protein side chains. Cys 25, with the 5-maleimidoproxyl label bound to it, is enhanced with dark ball-and-stick representation. The figure is created using Insight II.
Next, f-parameters were calculated for the minimized protein–lipid structures. In general, the lipid shell increased the f-parameter for residues 25, 31, 36, and 38 rather uniformly, whereas it had a discriminative effect at residues 46 and 49. For all mutants, the spin label was directed pointing away from the helix in the optimized structures (see Table 2). Exceptionally, this orientation produced an inconsistently high f-parameter for the maleimide connected to Cys 46, which is situated close to the lipid headgroups. However, for this particular residue, an orientation pointing outside the lipid shell (along the protein helix) produced a negligible change in energy. Therefore, the label on Cys 46 was fixed manually in the latter orientation.
Table 2.
Orientation of the spin-label N-O bond to the axis of the transmembrane helix, for structure number 20 of the M13 coat protein (PDB: 2CPS) optimized with single cysteine-maleimide substitutions at different positions, in the absence and presence of the lipid shell
| Spin-labeled residue no. | Without lipid | With lipida |
| 25 | 67° | 51° |
| 31 | 104° | 111° |
| 36 | 107° | 95° |
| 38 | 100° | 97° |
| 46 | 77° | 40°b |
| 49 | 70° | 70° |
a Final structure optimized with +1 Å vertical shift of the protein.
b Adjusted manually, producing an isoenergetic structure (see text).
Sequence profiles were then calculated for the f-parameter, not only with the protein in the position used for the structural optimization, but also with the protein displaced vertically in 1-Å increments, without further optimization. The resulting f-parameter profiles were then fitted to that of the EPR outer hyperfine splittings by using an adjustable linear scaling factor, plus offset, for the latter. The dependence of the fitting errors on the vertical displacement of the protein is given in Figure 7 ▶. The minimum fitting error was not found for zero vertical displacement of the protein, but for a displacement by +2 Å. Therefore, optimization of the entire protein–lipid structure was repeated with different vertical displacements of the protein. A consistent minimum fitting error was obtained for the structure optimized with a vertical displacement of +1 Å. This represents a one-residue shift, or less, relative to the original EPR estimate. The resulting sequence profile of the f-parameter is compared with that of the EPR outer hyperfine splittings in Figure 8 ▶. It is seen that the packing parameters deduced from the final optimized structure reproduce the major features of the experimentally determined mobility profile rather well. Including lipids in the model considerably improves the agreement with experiment (cf. Fig. 5 ▶); the rms matching error is reduced from 0.86 to 0.32, in the presence of lipids.
Fig. 7.

Matching error between the sequence profiles of the 14N outer hyperfine splitting (Stopar et al. 1997) and f-parameter, as a function of vertical displacement of the M13 major coat protein structure, relative to that shown in Fig. 6 ▶, in a DOPC bilayer. The minimum sum of squares of the differences was obtained by linear scaling of the outer splitting profile (with offset) to that of the f-parameter. Solid symbols indicate that the protein structure was not reoptimized at the new vertical position; open symbols denote reoptimization of the structure at the shifted position. Positive shift means that the N-terminal helix is moved closer to the membrane.
Final model
In the final optimized model, residues Tyr 24 and Phe 45 are located in the (somewhat diffuse) regions of the lipid phosphates on either side of the bilayer membrane. Formation of H-bonds between the ɛ-amino groups of Lys 43 and Lys 44 and the carbonyl groups of the lipid fatty acid chains is suggested by the model. To some extent this resembles the snorkel effect (Monné et al. 1998). At the opposite side of the bilayer, the model places Trp 26 in a position where it can function as a membrane-anchoring residue (Schiffer et al. 1992). In the mutated structure, the ɛ-amino group of Lys 40 interacts with oxygens of the maleimide ring for the spin label attached to Cys 36. This interaction reduces the overall potential energy of the system by 20 kcal/mol (evaluated in Sculpt). At least in part, this may be the reason for the increased outer hyperfine splitting at Cys 36 reported in Stopar et al. (1996, 1997). Putative H-bonds between the N–H group of the indole ring and the carbonyl oxygens of the fatty acid chains in adjacent lipid molecules possibly contribute to immobilization of the spin label on Cys 25 in the mutated structure. The N-terminal helix of the final structure is oriented parallel to the membrane surface (see Fig. 6 ▶), consistent with solid-state NMR results on the closely related fd coat protein in oriented membranes (McDonnell et al. 1993). An interesting feature of the amphipathic N-terminal surface helix (residues 8–16) is its azimuthal orientation relative to the membrane surface. This differs somewhat between the NMR structures determined in DodPC and SDS micelles. As already noted, the EPR data from phosphatidylcholine bilayer membranes, although not referring specifically to the N-terminal structure, is consistent only with the SDS-family. The N-terminal helix is oriented with the face containing the charged and polar residues Lys 8 and Gln 15, and aromatic residue Phe 11, directed towards the membrane. Correspondingly, the opposite face containing alanine residues 9, 10, and 16, and Ser 13, is directed towards the aqueous phase (cf. Papavoine et al. 1994). Such an orientation may not be the optimal one, as one would expect the strongly hydrophobic Leu 14 to be oriented toward the membrane rather than equatorially as it is in the model. This orientation of the N-terminal helix is likely to be specific for the strongly negative surface potential of SDS micelles and may be relieved by a slight twist of the helix with respect to the membrane in zwitterionic lipids. The final model remains hypothetical both in the sense that the starting structures are for a micelle rather than a bilayer environment and more particularly that only one coarse constraint has been used to distinguish between the N-terminal starting structures.
Conclusions
Relatively coarse-grained site-directed spin-label measurements have provided sufficient experimental constraints to select a single structural subclass from the family of high-resolution NMR structures in micelles as being that most appropriate to the M13 coat protein in lipid bilayer membranes. This has been achieved by using molecular mechanics optimization to model the spin-labeled protein in the presence of lipids. A relatively simple indicator of the local packing density (the f-parameter), which is readily calculated from the coordinates of the optimized protein–lipid structural model, was found to be adequate for this purpose. Extension of the approach to sparse experimental data on site-directed mutagenesis of other membrane proteins should be possible in the future. In our protein–lipid model, Trp 26 on one side of the membrane, and Lys 43 and Lys 44 on the other side, interact preferentially with the lipid head groups. The model indicates a hydrophobic mismatch of 3.5 Å or less (the protein is slightly shorter) between the unperturbed phospholipid bilayer and the intramembranous α helix of the protein. Spin-labeled Cys 25 is buried inside the hinge region, whereas Cys 46 points towards the aqueous phase, in agreement with their strong and weak motional restriction, respectively. Further, Cys 36 seems to be restricted by involvement in hydrogen bonding with Lys 40 in addition to steric hindrance from Val 33 and Ile 39. The model should prove useful for the interpretation of future experimental data on membrane–M13 major coat protein systems. It is also a good starting point for full-scale molecular dynamics simulations and for the design of further site-specific spectroscopic experiments.
Materials and methods
M13 major coat protein structures
Three-dimensional structures of the M13 major coat protein were taken from the Brookhaven Protein Data Bank in PDB format. The entry codes were 2CPB and 2CPS for structures determined in DodPC and SDS micelles, respectively, by various high-resolution NMR techniques (Papavoine et al. 1998).
Experimental data
EPR outer hyperfine splittings and membrane topology data for the viable single cysteine mutants A25C, V31C, T36C, G38C, T46C, and A49C of the M13 major coat protein reconstituted in DOPC bilayers were taken from (Stopar et al. 1997).
Molecular modeling
Recent versions of the quantum chemistry and molecular mechanics package Spartan (Wavefunction Inc., Irvine, CA) with MMFF94 force field, and the interactive molecular mechanics package Sculpt (Interactive Simulations Inc., San Diego, CA) with MM3 force field, were used for building and optimization of structures. Specifically, Spartan was used to generate the spin-labeled cysteine residue (validated by semi-empirical quantum chemical methods) and the phospholipid structure, and for reoptimization of the protein structure after single-residue replacement by spin-labeled cysteine. Additionally, Spartan was used for single-point energy calculations to obtain atomic charges. Adjustment of the phospholipid chain configuration and constrained molecular mechanics optimization of the protein–lipid assemblies were performed in Sculpt. MOLMOL (Koradi et al. 1996) was used for producing single-residue substitutions, construction of the lipid shell, and preparing the system for optimization by molecular mechanics. Insight II (Molecular Simulations Inc., San Diego, CA) was used for visualization and presentation of structures. All modeling work was performed on a Silicon Graphics (Mountain View, CA) Origin 2000 server and O2 workstations.
Electronic supplemental material
Atomic coordinates of the final optimized model, with the 5-MSL spin label bound on Cys 25, together with a single shell of DOPC lipids, is provided as exported from Sculpt (file name is m13lipid.xyz). The structure is similar to that presented in Figure 6 ▶, but was reoptimized after the protein was shifted by +1 Å relative to the lipid shell. This provided the lowest matching error (cf. Figs. 7,8 ▶ ▶). The data format follows that of the Protein Data Bank.
Acknowledgments
This work was supported by the Volkswagen-Stiftung (Germany), by the Hungarian National Science Foundation (OTKA T029458), and by the János Bolyai Foundation (Hungary). One of the SGI workstations for the modeling calculations was donated via the Max-Planck Gesellschaft (Germany).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
DOPC, dioleoylphosphatidylcholine
EPR, electron paramagnetic resonance
NMR, nuclear magnetic resonance
DodPC, dodecylphosphocholine
SDS, sodium dodecyl sulphate
5-MSL, 5-maleimidoproxyl
Supplemental material: See www.proteinscience.org
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.43801.
References
- Hemminga, M.A., Sanders, J.C., Wolfs, C.J.A.M., and Spruijt, R.B. 1993. Lipid-protein interactions involved in bacteriophage M13 infection. In Protein-lipid interactions, new comprehensive biochemistry 25 (ed. A. Watts), pp. 191–212. Elsevier, Amsterdam.
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 51–55. [DOI] [PubMed] [Google Scholar]
- Marsh, D. 1981. Electron spin resonance: Spin labels. In Membrane spectroscopy. Molecular biology, biochemistry and biophysics, Vol. 31 (ed. E. Grell), pp. 51–142. Springer-Verlag, New York. [DOI] [PubMed]
- Marsh, D. and Horváth, L.I. 1989. Spin-label studies of the structure and dynamics of lipids and proteins in membranes. In Applications in biology and biochemistry advanced EPR (ed. A.J. Hoff), pp. 707–752. Elsevier, Amsterdam.
- Marvin, D.A. and Hohn, B. 1969. Filamentous bacterial viruses. Bacteriol. Rev. 33 172–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin, D.A., Hale, R.D., Nave, C., and Citterich, M.H. 1994. Molecular models and structural comparisons of native and mutant class I filamentous bacteriophages Ff (fd, f1, M13), If1 and Ike. J. Mol. Biol. 235 260–286. [DOI] [PubMed] [Google Scholar]
- McDonnell, P.A., Shon, K., Kim, Y., and Opella, S.J. 1993. fd coat protein structure in membrane environments. J. Mol. Biol. 233 447–463. [DOI] [PubMed] [Google Scholar]
- Monné, M., Nilsson, I., Johansson, M., Elmhed, N., and von Heijne, G. 1998. Positively and negatively charged residues have different effects on the position in the membrane of a model transmembrane helix. J. Mol. Biol. 284 1177–1183. [DOI] [PubMed] [Google Scholar]
- Papavoine, C.H.M., Konings, R.N.H., Hilbers, C.W., and van de Ven, F.J.M. 1994. Location of the M13 coat protein in sodium dodecyl sulphate micelles as determined by NMR. Biochemistry 33 12990–12997. [DOI] [PubMed] [Google Scholar]
- Papavoine, C.H.M., Christiaans, B.E.C., Folmer, R.H.A., Konings, R.N.H., and Hilbers, C.W. 1998. Solution structure of the M13 major coat protein in detergent micelles: A basis for a model of phage assembly involving specific residues. J. Mol. Biol. 282 401–419. [DOI] [PubMed] [Google Scholar]
- Pascher, I. 1996. The different conformations of the glycerol region of crystalline acylglycerols. Curr. Opin. Struct. Biol. 6 439–448. [DOI] [PubMed] [Google Scholar]
- Rasched, I. and Oberer, E. 1986. Ff coliphages: Structural and functional relationships. Microbiol. Rev. 50 401–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer, M., Chang, C.H., and Stevens, F.J. 1992. The functions of tryptophan residues in membrane proteins. Protein Eng. 5 213–214. [DOI] [PubMed] [Google Scholar]
- Stopar, D., Spruijt, R.B., Wolfs, C.J.A.M., and Hemminga, M.A. 1996. Local dynamics of the M13 major coat protein in different membrane-mimicking systems. Biochemistry 35 15467–15473. [DOI] [PubMed] [Google Scholar]
- Stopar, D., Jansen, K.A.J., Páli, T., Marsh, D., and Hemminga, M.A. 1997. Membrane location of spin-labeled M13 major coat protein mutants determined by paramagnetic relaxation agents. Biochemistry 36 8261–8268. [DOI] [PubMed] [Google Scholar]
- Surles, M.C., Richardson, J.S., Richardson, D.C., and Brooks, Jr., F.P. 1994. Sculpting proteins interactively: Continual energy minimisation embedded in a graphical modelling system. Protein Sci. 3 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tristram-Nagle, S., Petrache, H.I., and Nagle, J.F. 1998. Structure and interactions of fully hydrated dioleoylphosphatidylcholine bilayers. Biophys. J. 75 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turyna, B., Osyczka, A., Kostrzewa, A., Blicharski, W., Enghild, J.J., and Froncisz, W. 1998. Preparation and electron paramagnetic resonance characterisation of spin labeled monoderivatives of horse cytochrome c. Biochim. Biophys. Acta 1386 50–58. [DOI] [PubMed] [Google Scholar]
- Van Wezenbeek, P.M.G.F., Hulsebos, T.J.M., and Schoenmakers, J.G.G. 1980. Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: comparison with phage fd. Gene 11 129–148. [DOI] [PubMed] [Google Scholar]
- Wolkers, W.F., Spruijt, R.B., Kaan, A., Konings, R.N.H., and Hemminga, M.A. 1997. Conventional and saturation-transfer EPR of spin-labeled mutant bacteriophage M13 coat protein in phospholipid bilayers. Biochim. Biophys. Acta 1327 5–16. [DOI] [PubMed] [Google Scholar]